
Abstract
Immunodeficiency-associated primary CNS lymphoma (PCNSL) represents a distinct clinicopathological entity, which is typically Epstein-Barr virus-positive (EBV+) and carries an inferior prognosis. Genetic alterations that characterize EBV-related CNS lymphomagenesis remain unclear precluding molecular classification and targeted therapies. In this study, a comprehensive genetic analysis of 22 EBV+ PCNSL, therefore, integrated clinical and pathological information with exome and RNA sequencing (RNASeq) data. EBV+ PCNSL with germline controls carried a median of 55 protein-coding single nucleotide variants (SNVs; range 24–217) and 2 insertions/deletions (range 0–22). Genetic landscape was largely shaped by aberrant somatic hypermutation with a median of 41.01% (range 31.79–53.49%) of SNVs mapping to its target motifs. Tumors lacked established SNVs (MYD88, CD79B, PIM1) and copy number variants (CDKN2A, HLA loss) driving EBV− PCNSL. Instead, EBV+ PCNSL were characterized by SOCS1 mutations (26%), predicted to disinhibit JAK/STAT signaling, and mutually exclusive gain-of-function NOTCH pathway SNVs (26%). Copy number gains were enriched on 11q23.3, a locus directly targeted for chromosomal aberrations by EBV, that includes SIK3 known to protect from cytotoxic T-cell responses. Losses covered 5q31.2 (STING), critical for sensing viral DNA, and 17q11 (NF1). Unsupervised clustering of RNASeq data revealed two distinct transcriptional groups, that shared strong expression of CD70 and IL1R2, previously linked to tolerogenic tumor microenvironments. Correspondingly, deconvolution of bulk RNASeq data revealed elevated M2-macrophage, T-regulatory cell, mast cell and monocyte fractions in EBV+ PCNSL. In addition to novel insights into the pathobiology of EBV+ PCNSL, the data provide the rationale for the exploration of targeted therapies including JAK-, NOTCH- and CD70-directed approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary central nervous system lymphoma (PCNSL) represents a rare extranodal variant of non-Hodgkin lymphoma (NHL) with an incidence of 0.45/100,000 [49]. Per definition, the disease is restricted to the central nervous system (CNS) at diagnosis including the brain parenchyma, spinal cord, meninges and eyes [31, 57].
Primary (e.g., genetic syndromes) or secondary (e.g., administration of immunosuppressants, human immunodeficiency virus (HIV) infection) immunodeficiency increases the risk of CNS lymphomagenesis and accounts for 5–10% of cases. These immunodeficiency-associated PCNSL constitute a distinct clinicopathological entity, which was recognized in the 2021 World Health Organization classification of CNS tumors [39]. Unlike disease in immunocompetent hosts, cases are typically (85–95%) Epstein-Barr virus-positive (EBV+), suggesting an EBV-related pathogenesis, and remain associated with an inferior outcome [9, 15, 26, 33, 47]. This may reflect either the inability to tolerate high-dose methotrexate-based polychemotherapy and/or EBV-driven oncogenesis with peculiar molecular properties. A better pathogenetic understanding of EBV+ PCNSL is hence necessary to pave the way for novel targeted therapies.
In recent years sequencing studies have helped to uncover the genetic landscape of immunocompetent, EBV− PCNSL [7, 11, 20, 30, 32, 50, 67]. Tumors predominantly harbor alterations in the B-cell receptor (BCR), toll-like receptor (TLR), and nuclear factor kappa B (NF-kB) signaling pathways. This includes PIM1, MYD88 L265P and CD79B Y196 hotspot mutations detected in 50–100%, 50–85% and 36–63% of cases, respectively. Genetics and tumor microenvironment of 44 EBV+ PCNSL were recently assessed using targeted sequencing and digital multiplex gene expression [21]. Tumors lacked hotspot mutations typically found in EBV− disease and were characterized by elevated macrophage and immune-checkpoint gene expression in HIV-negative and low CD4 gene counts in HIV-infected individuals. While this suggested a distinct molecular landscape, alterations that characterize EBV-related CNS lymphomagenesis remained unclear precluding further molecular classification and development of targeted therapies.
In this study, a comprehensive genetic analysis of 22 EBV+ PCNSL was therefore performed, which integrated clinical and pathological information with whole exome (WES) and RNA sequencing (RNASeq) data. This allowed us to uncover the protein-coding genetic landscape of EBV+ PCNSL and led to the identification of alterations, that may guide targeted treatment approaches of this rare entity.
Methods
Cohort and sample collection
Retrospective case review identified 19 treatment-naïve immunocompromised adult EBV+ PCNSL, collected at the Department of Neuropathology of University Hospital Heidelberg, with sufficient tissue for molecular characterization. Steroids were avoided in the two weeks before surgical diagnosis. Diagnosis was re-confirmed by a board-certified neuropathologist in all cases prior to study entry. Per histomorphological assessment, the tumor cell content exceeded 60% in all samples. Slit lamp examination and lumbar punctures were negative for ocular and cerebrospinal fluid involvement at diagnosis. Systemic disease was ruled out using fluorodeoxyglucose positron emission tomography or whole-body computed tomography as well as bone marrow biopsy. Retrospective chart review provided additional clinical and radiological data.
The discovery WES cohort included five fresh-frozen (FF) EBV+ samples with matching blood (germline) controls. Given the rarity of the disease, this cohort was supplemented with analyzed genetic data from three previously reported EBV+ cases that had matching blood controls (YNHH-6, LS-GD-0111, LS-GD-0106) [21, 32]. The in-house WES extension cohort consisted of FF or formalin-fixed paraffin-embedded (FFPE) tissue from 11/14 EBV+ samples, where matching blood controls were unavailable. Additionally, WES was carried out on two random EBV− PCNSL to confirm our pipeline detected previously established alterations. RNA sequencing (RNASeq) was performed on 19/19 EBV+ specimen from our institution, including all 16 in-house cases that also underwent WES.
Immunohistochemistry
Immunohistochemical stainings were performed on a Ventana BenchMark ULTRA Immunostainer (Ventana Medical Systems, Tucson, USA) using FFPE sections. The panel included CD20, CD10, BCL6, CD3, MUM1/IRF4, LMP1, PD-L1 and Ki67. Details on antibodies are provided in the supplementary. Samples were classified into germinal center B-cell-like (GCB) and non-GCB subtypes according to the Hans algorithm (BCL6, CD10, MUM1) [29].
EBV status assessment
EBV status was evaluated with immunohistochemistry (EBV-LMP1) and Epstein-Barr encoding region (EBER) in-situ hybridization following previously described protocols [16, 21]. Following previous studies, only cases where EBV was detected in more than half of the tumor cells were included in this study.
DNA and RNA extraction
Genomic DNA and RNA were extracted using the Promega Maxwell RSC device (Promega, Madison, USA). DNA was isolated with the Maxwell 16 Tissue DNA purification kit, the Maxwell 16 blood purification kit, or the Maxwell 16 FFPE Plus LEV DNA purification kit (Promega, Madison, USA) according to the manufacturer’s instructions. RNA was extracted with the Maxwell 16 LEV simplyRNA or the Maxwell 16 LEV RNA FFPE purification kit (Promega, Madison, USA) following the manufacturer’s protocols. Agilent 4200 TapeStation (Agilent Technologies, Santa Clara, USA) was used to determine RNA integrity numbers.
Whole exome sequencing and processing
Exome capture was performed with the Agilent SureSelect All Exon V7 kit (Agilent Technologies, Santa Clara, USA). The libraries were paired-end sequenced on an Illumina NovaSeq 6000 or NextSeq 500 (Illumina, San Diego, USA).
Alignment of reads
Mapping and preprocessing were performed in the DKFZ OTP (One Touch Pipeline) system using DKFZ AlignmentAndQCWorkflow version 1.2.73-202 (https://github.com/DKFZ-ODCF/AlignmentAndQCWorkflows). Briefly, reads were mapped to the human reference genome build hs37d5 (phase II reference of the 1000 Genomes Project including decoy sequences) using the Burrows-Wheeler Aligner (BWA) version 0.7.15 mem function with all default parameters, except minimum base quality threshold which was set to zero (−T 0) [5, 35]. BAM files were sorted using SAMtools version 0.1.19, and PCR duplicates were marked using Sambamba version 0.6.5 [36, 63].
SNVs
Single nucleotide variants (SNVs) were detected using an in-house workflow (https://github.com/DKFZ-ODCF/SNVCallingWorkflow) based on SAMtools/BCFtools with parameter adjustments to allow for somatic variant calling and heuristic filtering as previously described in the ICGC Pan-Cancer Analysis of Whole Genome project [65]. For paired samples, SNV calling was performed in OTP using SNVCallingWorkflow version 1.2.166-3 based on SAMtools/BCFtools version 0.1.19. For tumor samples without matched controls, SNVCallingWorkflow version 2.1.1-0 based on SAMtools/BCFtools version 1.9 was run in a no-control mode. No-control mode strategy included additional filters which removed common SNPs and recurrent artifacts using variant frequency information from public and local control sample pools. The local control pool contained variant frequency from 4879 whole genome (WGS) and 1198 whole exome (WES) samples analyzed with the same workflows. Variants with minor allele frequency (MAF) above 0.01 in 1000 Genomes or 0.001 in gnomAD (WGS or WES) or with a frequency above 0.01 in the local control pool (WGS or WES) were annotated as common SNPs or artifacts and removed from the downstream analysis [5]. To remove artifacts a 'confidence score' was calculated for each mutation. This score was initially set to 10 and subsequently reduced if the mutation overlapped with genomic regions that are known to be prone to artifacts. Those regions have been identified using the following UCSC genome browser tracks (http://genome.ucsc.edu/cgi-bin/hgTables [52]): GENCODE Mappability track, UCSC27 High Seq Depth track, UCSC Simple-Tandemrepeats, UCSC Repeat-Masker, DUKE-Excluded, DACBlacklist, UCSC Selfchain. High-confidence SNVs were defined as having a score of at least 8. Variants were annotated with GENCODE v19 using ANNOVAR (version 2016Feb01) [18, 70]. Only somatic non-silent coding variants (i.e., nonsynonymous, stopgain, stoploss, or splicing in a vicinity of 2 bp of exon boundaries) of high confidence were selected (except for the analysis of mutational signatures, where all high confidence, including non-coding and silent, somatic variants from the capture regions were used).
INDELs
Short insertions/deletions (indels) were identified using in-house workflow (https://github.com/DKFZ-ODCF/IndelCallingWorkflow) based on Platypus (version 0.8.1.1) as previously described [65]. For paired samples, indel calling was performed in OTP using IndelCallingWorkflow version 2.4.1-1. For tumor samples without matched controls, IndelCallingWorkflow version 3.1.0-0 was run in a no-control mode. As in the SNV workflow, the no-control mode strategy included additional filters based on variant frequency information from public and local control sample pools. A scoring scheme like that applied in the SNV workflow was used. Indels were annotated with GENCODE v19 using ANNOVAR and somatic high-confidence indels (minimum confidence of 8) falling into a coding sequence or splice site were selected for the analysis.
Copy number alterations (CNAs)
Copy number alterations (CNAs) and loss of heterozygosity regions of in-house samples were identified using a workflow based on the software package CNVkit version 2.1.0 with default parameter setting [61]. For tumor samples without matched controls, same-gender control samples from other patients were used as “artificial controls”. Tumor cell content and ploidy were estimated using a method adapted from ACEseq (https://aceseq.readthedocs.io/en/latest/. Segments with a total copy number (TCN) at least 0.7 above the tumor ploidy were defined as gains and segments with a TCN at least 0.7 below it were defined as losses. High-level CNAs were defined as homozygous deletions and amplifications (> 2.5 × the average ploidy). CNA analysis in FFPE samples is challenging and may result in artifacts, particularly with respect to copy number losses. To address this, GISTIC2.0 workflow with default parameters (except for q = 0.1 and log2 ratio amplification/deletion threshold = 0.3/− 0.3) was used to identify regions of significantly recurring gains or losses [44]. Segmented log2 ratios (.cns) were generated by CNVkit and converted to a segmentation.seg file. This file was then used as input for the GISTIC2.0 module.
Mutational signature
Mutational signature contributions were calculated using the R/Bioconductor package YAPSA (Yet Another Package for Signature Analysis, https://bioconductor.org/packages/release/bioc/html/YAPSA.html); the Alexandrov-COSMIC (AC) signatures (https://cancer.sanger.ac.uk/cosmic/signatures_v2) [2, 3].
Viral integration detection
The presence of viral DNA and genomic integration in tumor samples was detected using an in-house pipeline based on VIRUSBreakend [8]. Starting with an aligned BAM file, it identifies viral reads of interest through Kraken2 taxonomic classification of all unaligned or partially aligned sequences using a custom Kraken2 database [73].
RNA sequencing (RNASeq)
Sequencing, processing, and alignment
Illumina TruSeq stranded mRNA kit was used to prepare RNASeq libraries. Sequencing was performed on the Illumina HiSeq X Ten V2.5 platform (Illumina, San Diego, USA). Paired-end reads were aligned to the human genome (hg19) using STAR (v2.5.2b) with default settings. Sambamba (v0.4.6) marked duplicate reads. SAMtools (v1.19) allowed coordinate sorting of BAM files. Gene-specific reads were quantified with the human gene model (GENCODE v19) using featureCounts in Subread (v1.5.1) with default settings. Gene read count matrix was obtained for further analyses.
Unsupervised clustering analysis
The top 500 highly variable genes were selected for hierarchical clustering analysis. The optimal number of clusters in hierarchical clustering was selected based on the Silhouette method using R package factoextra (v1.0.7). Three clusters were obtained (i.e., EBV_1, EBV_2 and EBV_3).
Differential expression (DE) analysis
Pairwise DE analyses between three clusters (i.e., EBV_1 vs. EBV_2, EBV_2 vs. EBV_3 and EBV_1 vs. EBV_3) were performed in R package DESeq2 (v1.32.0) [40]. Genes with total counts < 10 in all samples were excluded. Genes were normalized with variance stabilizing transformation (vst) method. DE results were shrunk using apeglm. The top 50 DE genes (DEGs) in comparisons with adjusted p value < 0.05 and log2 fold change > 2 (upregulated DEGs) or log2 fold change < 2 (downregulated DEGs) were selected as features for clusters. For example, EBV_1 included the top 50 upregulated DEGs and the top 50 downregulated DEGs in EBV_1 vs. EBV_2 and EBV_1 vs. EBV_3. Expression clusters were compared with the gene read count matrix of healthy brain tissue RNA-Seq data downloaded from the Genotype-Tissue Expression (GTEx) Portal (https://www.gtexportal.org/home/). The seven samples (Supplementary Table 5) were randomly selected and included various brain areas from both genders.
Gene ontology (GO) enrichment analysis
The enriched GOs for each cluster were obtained using R package clusterProfiler (v4.0.5) with biological process Gos from the org.Hs.eg.db database. The top 10 Gos with q value < 0.05 were visualized in bar plots using ggplot2 (v3.3.5).
Deconvolution of bulk RNA Sequencing
Deconvolution of bulk RNA Sequencing (counts-per-million [CPM] values) from our EBV+ and a previously published EBV− cohort from the ICGC-MML-Seq consortium [50] were performed using CIBERSORTx (https://cibersortx.stanford.edu) [48]. The signature matrix of human hematopoietic cell phenotypes (LM22) defined immune cell subsets [12]. The imputed cell fractions were visualized in box plots using ggpubr (v0.4.0).
Validation of sequencing
Selected variants (SOCS1, NOTCH1, KMT2D, IGH-V169) were validated using Sanger Sequencing of tumor and blood control samples. For validation of copy number variants (CNVs) and RNASeq data we subjected genomic and synthesized complementary DNA to quantitative PCR, respectively. Details are provided in the supplementary.
Ethics
This study was approved by the Heidelberg University Medical Faculty institutional review board (S-128/2022). Written informed consent was obtained from all study participants or their legal guardians. The study was conducted in accordance with the Declaration of Helsinki.
Results
Cohort characteristics
The study cohort included 8 paired and 14 unpaired treatment-naïve immunodeficiency-related EBV+ PCNSL. All cases were classified as diffuse large B-cell lymphoma (DLBCL). The cell-of-origin was determined according to the Hans algorithm with most lymphomas attributed to the ABC subtype (n = 20). Clinical/pathological cohort characteristics are summarized in Supplementary Table 1.
Exome sequencing
Exome sequencing was performed with a median coverage of 226X (range 41–526) and 294X (range 248–321) on in-house tumor and blood specimen, respectively. Sequencing data was assessed for the presence of viral genetic material. EBV sequences (NC_007605.1) were detected in all included EBV+ cases, matching pathological evaluations. No overlap with viral sequences was found in both EBV− tumors (Supplementary Fig. 1), that carried established SNVs (e.g., CD79B p.Y196H, PIM1, TBL1XR, HLA-B, HLA-C) and CNVs (e.g., CDKN2A and HLA loss).
Exonic variant counts
In the paired EBV+ PCNSL samples (n = 8, Fig. 1a), a median of 55 functional, protein-coding SNVs (range 24–217) and a median of 2 INDELs (range 0–22) were found. Base transitions revealed a C > T predominance across samples in agreement with deaminated unmethylated cytosines (Fig. 1c). Median tumor mutational burden (TMB) was 1.9/megabase (Mb) (Fig. 1b). Comparison to other tumor entities deposited in the cancer genome atlas (TCGA) revealed median TMB in immunocompetent systemic DLBCL patients was higher (3.8/Mb). Similarly, according to a recently published dataset [50], TMB in EBV− PCNSL (7.5/Mb) exceeded EBV+ counterparts included in this study. This was reflected by a significantly higher number of functional exonic SNVs (p < 0.001, Fig. 1d) and INDELs (p < 0.01, Fig. 1e) in EBV− PCNSL.

Single nucleotide variant and insertion/deletion counts. a Functional exonic single nucleotide variants (SNVs, blue) and insertions/deletions (INDELs, yellow) counts are displayed. Paired EBV+ PCNSL carried a median of 55 protein-coding SNVs (range 24–217) and 2 INDELs (range 0–22). b Tumor mutational burden (TMB: variants/megabase (Mb)) for various malignancies deposited in the cancer genome atlas (TCGA) are shown and contrasted to EBV+ PCNSL from this study and EBV− counterparts from the ICGC MMML-Seq consortium. TMB in EBV+ PCNSL was lower than in systemic lymphomas (DLBCL) and EBV− PCNSL. c Base substitutions were conserved across cases and indicate a C > T predominance in agreement with the deamination of cytosines. d EBV+ PCNSL carried significantly (Mann–Whitney test; ***p < 0.001; **p < 0.01) fewer non-synonymous exonic SNV and INDELs (e) than EBV− PCNSL from the ICGC MMML-Seq consortium
Aberrant somatic hypermutation
Genetic landscape of EBV− PCNSL is largely shaped by aberrant somatic hypermutation (aSHM), which is driven by activation-induced cytidine deaminase (AID) enzyme-mediated cytosine deamination [32, 50, 67]. It targets the WRCY sequence motif (W = A/T; R = A/G; C = hotspot; Y = C/T) and its reverse complement, RGYW. To assess the relevance of aSHM in EBV+ disease, the fraction of SNVs that overlapped with its target motifs was determined in our paired cohort (Fig. 2a). A median of 41.01% (range 31.79–53.49%) of SNVs localized to the WRCY (median: 17.5%, range 8.16–25.58%) or RGYW (median 21.94%, range 15–40.82) motifs. The WRCY and RGYW motif hotspots were targeted in 4.41% (range 2.04–9.3%) and 7.28% (range 3.33–18.37%), respectively. Despite the high overlap of SNVs with aSHM motifs, paired EBV+ tumors lacked PIM1 variants, the most prominent target of aSHM in EBV− PCNSL [50, 67]. In addition to somatic hypermutation of immunoglobulin genes (Fig. 2b, c), SOCS1 SNVs frequently mapped to aSHM motifs (Fig. 2d) and were found within the first 2000 bases from the transcriptional start site.

Aberrant somatic hypermutation EBV+ PCNSL. a SNV fractions that overlapped with aberrant somatic hypermutation target motifs WRCY (yellow) and RGYW (blue) were determined (W = A/T; R = A/G; C or G = hotspot; Y = C/T). A median 41.01% of SNVs localized to either motif, suggesting a critical role for aSHM in the pathobiology of EBV+ PCNSL. b, c, d Genes with alterations within the first 2000 (K = 1000) bases from the transcriptional start site, that frequently mapped to aSHM motifs, are shown. Positions of detected variants are indicated with symbol shapes denoting relation to target motifs. Targets in EBV+ PCNSL included immunoglobulin genes (b, c), but also SOCS1 (d)
Altered genes in EBV+ PCNSL
Detected SNVs were filtered for relevance in tumorigenesis (Fig. 3a) using the COSMIC Gene Census database and the network of cancer genes catalogue of known and candidate cancer genes (NCG.v7). Immunoglobulin gene SNVs, which constitute a feature of healthy and neoplastic B-cells resulting from somatic hypermutation, were found in all cases. EBV+ PCNSL lacked known MYD88 p.L265P and CD79B p.Y196 hotspot mutations, which were previously described to drive CNS lymphomagenesis in EBV− cases (Fig. 3b) [7, 11, 20, 30, 32, 50, 67]. PIM1 and TBLXR1, two frequently altered genes in EBV− PCNSL such as in our control samples, were altered in a single unpaired case only (HD-272204). Overall, except for the above-mentioned case, EBV+ PCNSL harbored few alterations in the BCR and NF-kB signaling pathways.

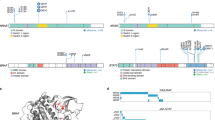
Single nucleotide variants in EBV+ PCNSL. a Filtered genes with functional exonic SNVs and INDELs are shown. Colors indicate the type of alteration. SOCS1 was the most frequently (26%, n = 5) altered gene in EBV+ PCNSL. Mutually exclusive NOTCH variants were detected in 26% (n = 5) of tumors. b Frequencies of selected alterations are contrasted to EBV− PCNSL (blue) from the ICGC-MMML-Seq dataset. EBV+ cases (red) mostly lacked MYD88, CD79B, and PIM1 variants characteristic of virus-negative disease (blue). Frequencies were compared with Fisher Exact tests and significance levels are displayed (****p < 0.0001; ns = not significant). c Normalized single base substitution Alexandrov-COSMIC (AC) mutational signatures in EBV+ PCNSL are shown. Colors indicate the signature type and proportions reflect related SNVs counts/sample. Signatures associated with spontaneous deamination of cytosines (AC1), and defective DNA mismatch repair (AC20, AC21) were frequently found. d The SOCS1 gene and its domains are displayed (KIR, kinase inhibitory region; ESS, extended SH2 subdomain; SH2, Src2 homology domain; SOCS box) and detected alterations are indicated. Most (n = 10/13) variants clustered to the SH2 domain, which mediates binding and subsequent inhibition of JAK1. e Crystal structure of the SOCS1 SH2 domain (red) in complex with the JAK1 kinase domain (green) obtained from the RCSB protein data bank (accession number 6C7Y). Positions of altered amino acids (blue) in EBV+ PCNSL are indicated within the complex
JAK/STAT pathway
As a target of aSHM, the most frequently altered gene in EBV+ PCNSL was SOCS1 with heterozygous variants detected in five cases (26%) including two with germline controls (Fig. 3a). Most hits (n = 10/13) clustered within the SH2 domain (Fig. 3d, e), which mediates binding to JAK1, critical for inhibition of the JAK/STAT pathway [38]. This included recurrent variants p.Y80F (n = 3), p.G88A (n = 2), and p.A89P (n = 2). Several alterations (p.N11T, p.A17V, p.W81R, p.G88A, p.A89P, p.S116R) were annotated in the COSMIC database and previously reported in systemic DLBCL. MutationTaster predicted p.N11T, p.Y80F, p.W81R, p.A89P, p.S116R, and p.I126M were deleterious. Bioinformatic loss-of-function predictions were additionally corroborated by two samples carrying multiple non-synonymous SNVs and matched previously suggested tumor suppressive properties of SOCS1 in lymphoma.
Heterozygous germline SOCS1 variants were previously linked to severe autoimmunity with aberrant lymphoproliferation and transition to lymphomas in some cases [27]. Blood controls from this study were therefore assessed for additional SOCS1 alterations. Second hits in cases with somatic SNVs were absent. However, a germline SOCS1 variant of unknown significance (p.Q210H, rs11549428) was found in one individual with autoimmune disease-related PCNSL. This variant is found at a population frequency of 0.18% according to the 1000 Genomes Project, lies outside the SH2 domain and could also represent a benign polymorphism.
Somatic variants were detected in additional inhibitory JAK/STAT pathway genes, namely SOCS gene family member SOCS6 (p.K65I) as well as adaptor protein SH2B3 (p.D69N). MutationTaster predicted deleterious effects for variants further suggesting disinhibition of the JAK/STAT pathway.
NOTCH pathway
NOTCH SNVs occurred in five EBV+ PCNSL (26%). NOTCH1 variants were uncovered in three specimens (16%) including one paired case (Fig. 3a). Two of three variants localized to heterodimerization (p.R1598H) and PEST (p.H2500L) domains, respectively, that carry known hotspots for gain-of-function mutations [71]. SNVs in the heterodimerization domain were previously shown to expose the metalloprotease S2 cleavage site resulting in ligand-independent cleavage, and release of the intracellular receptor domain with subsequent pathway activation. Alterations in the PEST domain prevent ubiquitin-mediated proteasomal degradation of the activated receptor. MutationTaster prognosticated pathogenic consequences for detected NOTCH1 SNVs. Additional hits in the NOTCH pathway included NOTCH3 (n = 2, 11%) and NOTCH4 variants (n = 1, 5%). NOTCH3 p.G1422A was found in the proximity of the heterodimerization domain whereas NOTCH4 p.R1924W localized to the PEST domain suggesting gain-of-function. Of note, NOTCH and SOCS1 SNVs were mutually exclusive in EBV+ PCNSL.
Epigenetic regulators, which are commonly mutated in EBV− disease, also carried several SNVs in EBV+ PCNSL (Fig. 3a). Overall, variants in respective genes were detected in eleven cases (58%). They co-occurred with JAK/STAT or NOTCH pathway alterations. Affected genes included lysine methyltransferases KMT2 genes (KMT2D n = 3; KMT2B n = 2; KMT2E: n = 2), DNA-methyl-transferase (DNMT) genes (DNMT1 n = 3; DNMT3A n = 2; DNMT3B n = 2), and lysine acetyltransferase CREBBP (n = 2) among others.
Mutational signatures
Detected variants were compared with previously described Alexandrov-COSMIC (AC) mutational signatures (Fig. 3c) [2, 3]. Patterns associated with spontaneous deamination of 5-methylcytosine (AC1), which is enriched in EBV− PCNSL [50], were frequently detected in EBV+ PCNSL. Variants were also linked to AID/APOBEC family of cytidine deaminases activity (AC2). Overlap with signatures related to defective DNA mismatch repair (AC20, AC21) and homologous recombination DNA mismatch repair (AC3) were frequently identified. Additionally, mutational patterns of unknown etiology were found (AC12, AC17).
CNAs
A median of 6.5% (range 2.5–14.7%) of the genome were affected by CNAs (Fig. 4). CDKN2A or HLA loss, characteristic for EBV− PCNSL, were not found EBV+ cases. Frequent gains were found on 11q23.3 (FOXR1, SIK3), a site known to be directly targeted for genomic aberrations by EBV protein, EBNA1 [37]. Additionally, gains included 21q22.2 (ERG) and NOTCH1. The latter were also found in samples, which lacked additional NOTCH pathway SNVs, providing an alternative mechanism of pathway activation. Losses were enriched on 5q31, the locus of STING1 critical for sensing viral DNA with subsequent induction of apoptosis [46]. Further losses were identified on 7q32 (IRF5), and 17q11 (NF1).

Copy number alterations in EBV+ PCNSL. Chromoplot summarizes copy number variants (CNVs) of in-house EBV+ PCNSL. Chromosomes are displayed on the x-axis. The G-score, which integrates frequency across samples and the magnitude of CNVs, is shown on the y-axis. Gains (red) and losses (blue) are indicated. Lymphomas carried 11q23, a site known to be specifically targeted for genomic aberrations by EBV, and 21q22 gains. Losses covered 5q31, the locus of STING1 necessary for sensing of viral cytoplasmic DNA, 7q32 and 17q11
RNA sequencing
Expression clusters
Unsupervised hierarchical clustering revealed three expression clusters in EBV+ PCNSL (Fig. 5a). As normal brain contamination was encountered in previous studies performing RNA-sequencing on PCNSL specimen [50], DEGs were compared with expression patterns from healthy brain controls deposited in GTEx. DEGs found in cluster 1 largely reflected brain tissue expression patterns. This was further corroborated by enriched gene ontology (GO) terms (Supplementary Fig. 3) and cases were excluded from further analyses. In contrast, clusters two and three shared B-cell related terms including ‘regulation of B-cell activation’, or ‘regulation of immune effector processes’ (Fig. 5b). Of note, cases with SOCS1 alterations were found within expression cluster 2, whereas tumors with NOTCH1 SNVs mapped to cluster 3.

Expression clusters and tumor microenvironment in EBV+ PCNSL. a Heatmap shows scaled expression levels of feature genes from three EBV+ PCNSL expression groups, identified with unsupervised hierarchical clustering analysis, across samples and healthy brain controls from the GTEx database. Cluster 1 (EBV_1) showed high overlap with GTEx controls in line with healthy brain contamination and cases were removed from further analyses. Clusters 2 (EBV_2) and 3 (EBV_3) separated lymphomas with SOCS1 and NOTCH1 SNVs and displayed robust IL1R2 and CD70 expression. b Top 10 enriched gene ontology (GO) terms for feature genes from these clusters are shown. GOs were ordered with rising q values from top to bottom and number (N) of cluster genes overlapping with respective GO terms are indicated along the x-axis. GOs largely reflected B-cell-related terms. c Heatmap indicates robust expression of immune checkpoint genes and markers of a tolerogenic tumor microenvironment (TME) across EBV+ PCNSL. d Correspondingly, deconvolution of bulk RNASeq data (CIBERSORTx) from EBV+ PCNSL and EBV− cases from the ICGC-MMML-Seq dataset revealed a tolerogenic TME with enrichment of T-regulatory cell, M2-macrophage, monocyte, and mast cell fractions in EBV-related tumors (Mann–Whitney test; ****p < 0.0001; ***p < 0.001, **p < 0.01, *p < 0.05; ns not significant)
Expression groups 2 and 3 shared robust IL1R2 and CD70 expression. Corresponding to strong expression in EBV+ PCNSL, analysis of the GTEx portal database revealed CD70 overexpression in EBV-transformed B-lymphocytes (median 340.6 transcripts per million, Supplementary Fig. 4). Significantly DEGs between both clusters included stronger immunoglobulin constant gene and MAPK4 expression in cluster 2 whereas TRAF4 and SMO were among upregulated genes in group 3 (Supplementary Fig. 5).
CIBERSORTx analysis suggests a tolerogenic tumor microenvironment
In addition to immune checkpoints (PD-L1/L2, LAG3, TIM-3) strongly expressed IL1R2 and CD70 can inhibit immune responses and create a tolerogenic tumor microenvironment (TME) [41, 64]. We found strong expression of CD163 (M2-macrophages) and FOXP3 (T-regulatory cells, Treg; Fig. 5c) across EBV+ PCNSL. To further characterize the TME, deconvolution of bulk RNA sequencing data was performed using CIBERSORTx (Fig. 5d) comparing our EBV+ dataset with previously published EBV− PCNSL [50]. In agreement with a tolerogenic TME significant enrichment of Tregs, M2-macrophages, monocytes and mast cells were found in EBV+ PCNSL.
Discussion
In this study, comprehensive genetic analyses of 22 EBV+ PCNSL integrated clinical and pathological data with WES and RNASeq. This provided novel mechanistic insights into EBV-driven CNS lymphomagenesis and revealed alterations, that may serve as diagnostic markers and/or may guide targeted therapy.
Previous work suggested EBV+ PCNSL lack hotspot mutations detected in EBV− PCNSL [11, 32]. This was further supported by a recent study [21], that characterized 44 EBV+ PCNSL FFPE samples using targeted sequencing (54 genes) and a gene expression panel (14 genes). In agreement with our WES findings, SNVs (PIM1, MYD88, CD79B) and CNVs (CDKN2A, HLA loss) characteristic of EBV− disease were absent. Corresponding to fewer alterations within their sequencing panel, WES now revealed a lower TMB in EBV+ PCNSL, which parallels other EBV+ lymphomas outside the nervous system [22]. However, alterations that drive EBV+ PCNSL remained unclear given their study design, and top hits from this WES/RNASeq study (Fig. 3 SNVs: SOCS1, NOTCH; Fig. 4 CNVs: 11q23.3 gain, 5q31.2 loss; Fig. 5 RNA: CD70, IL1R2) were not part of their panel.
The genetic landscape of NHL is largely shaped by aSHM driven by AID activity [32, 50, 67]. As EBV+ PCNSL lacked variants in most prominent targets including PIM1, its influence on the genetic landscape was unclear. Although target genes differed, the high overlap of SNVs with aSHM motifs, however, suggested a critical role during EBV-driven CNS lymphomagenesis. This mirrors other EBV-related tumors such as Burkitt lymphoma, where the genetic landscape is largely shaped by AID activity and subsequent aSHM [24]. Underlining its pathogenetic relevance in EBV+ PCNSL, like PIM1 in EBV− tumors the most frequently mutated gene in EBV+ disease, SOCS1, represents a known target of aSHM and several detected SNVs clustered within its target motifs [45].
The suppressor of the cytokine signaling (SOCS) gene family including its most potent member, SOCS1, provides an inducible negative feedback loop for cytokine-triggered JAK/STAT pathway activation [69]. SOCS1 directly interacts with JAK1 via its SH2 domain, which results in ubiquitination and subsequent proteasomal degradation of JAK1 as well as direct inhibition of Janus kinase activity [38]. Most detected variants in EBV+ PCNSL localized to the SH2 domain and bioinformatic scores predicted loss-of-function effects pointing to JAK/STAT pathway disinhibition. SOCS1 mutations were also documented in EBV− PCNSL albeit reported at lower frequencies [30, 50]. Whether SOCS alterations are indeed enriched in EBV+ disease warrants further investigation given the available case numbers. However, detection in other EBV-related lymphomas (systemic DLBCL including HIV-related disease; Hodgkin lymphoma) supports their relevance during EBV-driven lymphomagenesis [10, 22, 72]. Of note, JAK/STAT activation is known to drive the expression of EBV oncogenes including LMP1 via direct interaction of STAT molecules with promoter regions in EBV-related tumors [13, 14]. Additionally, the pathway up-regulates immune checkpoint PD-L1, strongly expressed in EBV+ PCNSL, and may hence contribute to a tolerogenic microenvironment [25]. SOCS1 variant-related disinhibition of the pathway may allow to unleash the viral oncogenic potential. Modeling of similar SOCS1 variants—including SH2 domain alterations—in EBV-transformed B-lymphocytes and HEK293 cells previously revealed excessive JAK/STAT pathway activation upon cytokine stimulation [27]. This sensitized for treatment with the JAK1/JAK2 inhibitor ruxolitinib, which is approved for the treatment of myeloproliferative neoplasms and was shown to penetrate the blood–brain barrier in mice [28, 68]. These findings provide a rationale for preclinical and early clinical assessment of JAK inhibition in EBV+ PCNSL.
In addition to JAK/STAT pathway variants, mutually exclusive NOTCH SNVs were detected in EBV+ PCNSL. Most alterations localized to mutational hotspots in the heterodimerization and PEST domains, which result in gain-of-function in T-cell and chronic lymphocytic leukemia [53, 71]. Additional NOTCH1 amplifications provide an alternative mechanism and further support genetic pathway activation in EBV+ PCNSL. This matches EBV+ systemic lymphomas, where activating NOTCH2 SNVs were detected [22, 58], which are rare in virus-negative tumors. NOTCH activation was previously shown to maintain oncogenic viral latency transcriptional programs and prevent lytic cycles and subsequent cell death in EBV+ tumors, which was reverted with NOTCH inhibitor treatment [23]. Additionally, NOTCH drives immune evasion via induction of PD-L1 and could contribute to a tolerogenic microenvironment in EBV+ PCNSL [42]. Various NOTCH inhibitors are under ongoing preclinical/clinical development and evaluation in EBV+ PCNSL may be warranted based on alterations identified in this study [4].
Frequent detection of variants in epigenetic regulators was in line with the previous targeted sequencing approach, which identified KMT2D/KMT2C as top hits in EBV+ PCNSL although mutational frequencies were lower than in EBV− tumors [21]. However, they were among the few typical variants shared between virus-positive and -negative lymphomas. KMT2D, the most frequently altered epigenetic modifier in EBV+ PCNSL, also represented the top hit in a CRISPR-Cas9 screen investigating DLBCL drivers [51]. This may suggest that epigenetic regulators are implicated in CNS lymphomagenesis irrespective of EBV status.
Distinct CNV profiles were identified in EBV+ PCNSL, which overall may impair antiviral defense mechanisms and promote B-cell activation. This included 11q23 gains, the locus of SIK3, which protects cells from cytotoxic T-cell responses [59]. Of note, this locus is specifically and directly targeted for genomic aberrations by the EBV protein, EBNA1 [37]. An 11q23 gain/loss pattern was previously found in posttransplant Burkitt-lymphoma-like disease [54], whereas tumors in immunodeficient hosts from this cohort lacked respective deletions. Additionally, loss of STING1 (5q31.2) may hamper cytoplasmic viral DNA sensing and disinhibit BCR signaling [46, 62, 74]. 7q32 deletions cover IRF5, a known tumor suppressor in B-cell lymphoma, while individuals with NF1 loss are at increased risk for NHL [6, 60].
Transcriptional profiling revealed two distinct clusters in EBV+ PCNSL, that separated cases with SOCS1 and NOTCH1 SNVs. In addition to previously reported robust PD-L1 expression [11, 21], both expression groups shared strong IL1R2 and CD70 expression, which may shape a tolerogenic TME. IL1R2 constitutes a decoy interleukin receptor, that abrogates interleukin-1 signaling and is known for strong expression in serum and tumor tissue in EBV-related Hodgkin lymphoma [41]. CD70 belongs to the TNF receptor superfamily and is predominantly expressed on activated B- and T-cells. Via interaction with its receptor, CD27, it acts as an immune checkpoint that can co-stimulate B-cell activation, but also inhibit immune responses via T-cell exhaustion/apoptosis [17, 64]. Overexpression in tumors was linked to more aggressive phenotypes and allowed immune evasion via a tolerogenic TME. Given the absence of expression in most healthy tissue, CD70 is evaluated as a novel immunotherapeutic target and CD70-directed antibodies as well as CAR T-cells showed promising efficacy in preclinical but also early clinical studies [17, 56]. This study revealed CD70 as an interesting candidate for targeted therapy in EBV+ PCNSL. Further membrane proteins that shared robust expression in EBV+ tumor samples and low/absent levels in most healthy tissues included PD-L1, PD-L2, LAG3, TIM3 as well as B-cell marker CD19. As a result, these genes carry the potential for immunotherapeutic approaches, some of which have already been explored in EBV− PCNSL as well as in other malignancies [19, 34]. However, immunocompromised patients were excluded from most clinical trials due to concerns for immune-related adverse events (irAE). While a growing body of evidence shows that immunotherapy can be safely administered in many immunodeficient patients and most irAE are manageable, an individual risk–benefit analysis as well as vigilant monitoring in a referral center setting are warranted [1, 43, 66].
In agreement with the overexpression of immune checkpoint genes, RNA-sequencing suggested a tolerogenic TME including the presence of Tregs (FOXP3) and particularly M2-macrophages (CD163). Similarly, CD163 was previously reported significantly up-regulated in EBV+ PCNSL compared to EBV− counterparts [21]. Deconvolution of bulk RNASeq datasets in this study further revealed higher M2-macrophages, Treg, monocyte and mast cell fractions in EBV+ PCNSL. This is in agreement with other EBV-related lymphomas (e.g. Hodgkin), where immune evasion via a tolerogenic TME contributes to virus-driven lymphomagenesis [55].
Limitations of this study include the cohort size, which reflects the rarity of EBV+ PCNSL and/or the lack of tissue availability. However, sequencing studies in more prevalent EBV− disease enrolled similar or even lower case numbers [7, 11, 20, 32, 67]. Germline controls were also not available for the entire cohort, which was addressed with a rigorous filtering approach based on recurrent and previously reported alterations. Supplemented, previously described paired cases were processed using different workflows, which will have introduced batch effects. Yet high similarity to in-house paired samples including detection of recurrent SOCS1 and NOTCH alterations additionally supported our findings. Strengths of this study include the integrated WES/RNASeq approach, which led to the identification of novel variants and markers in EBV+ PCNSL (e.g., SOCS1, NOTCH1, CD70) and suggested a tolerogenic TME. In addition to insights into the pathomechanism of this rare entity this provides the rational for the exploration and development of new targeted therapies. This is particularly important as many EBV+ patients are unable to tolerate aggressive methotrexate-based polychemotherapy resulting in an inferior outcome. Previous functional studies together with bioinformatic scores provide a good body of evidence for the likely functional relevance and/or targetability of identified alterations. Yet further in vitro and in vivo characterization should be considered in future studies to allow rapid translation of our findings from bench to bedside. Additionally, a future immunogenomics approach (e.g. HLA typing, neoepitope prediction with subsequent experimental validation) could allow us to identify further targets for immunotherapy.
Conclusions
The genetic landscape of EBV+ PCNSL is largely shaped by aSHM although tumors lacked variants characteristic of EBV− disease. Lymphomas frequently harbored SNVs in the JAK/STAT and NOTCH signaling pathways while CNVs point to impaired antiviral defense mechanisms. Transcriptional profiling revealed two distinct expression clusters, which shared robust expression of immune checkpoint genes including CD70 and IL1R2. Correspondingly deconvolution of bulk RNASeq data suggested a tolerogenic tumor microenvironment. Findings pave the avenue for the (pre-)clinical evaluation of several targeted therapies including JAK/STAT and NOTCH inhibition as well as CD70-directed treatments.
Data availability
WES and bulk RNASeq were deposited in European Genome-phenome Archive (accession: EGAS00001007222). Data access can be obtained from the listed Data Access Committee upon reasonable request. Remaining data are provided within this article and its supplementary. All codes are publicly available.
References
Abramson JS, Irwin KE, Frigault MJ, Dietrich J, McGree B, Jordan JT et al (2019) Successful anti-CD19 CAR T-cell therapy in HIV-infected patients with refractory high-grade B-cell lymphoma. Cancer 125:3692–3698. https://doi.org/10.1002/cncr.32411
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y et al (2020) The repertoire of mutational signatures in human cancer. Nature 578:94–101. https://doi.org/10.1038/s41586-020-1943-3
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV et al (2013) Signatures of mutational processes in human cancer. Nature 500:415–421. https://doi.org/10.1038/nature12477
Andersson ER, Lendahl U (2014) Therapeutic modulation of Notch signalling—are we there yet? Nat Rev Drug Discov 13:357–378. https://doi.org/10.1038/nrd4252
Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR et al (2015) A global reference for human genetic variation. Nature 526:68–74. https://doi.org/10.1038/nature15393
Barnes BJ, Kellum MJ, Pinder KE, Frisancho JA, Pitha PM (2003) Interferon regulatory factor 5, a novel mediator of cell cycle arrest and cell death. Cancer Res 63:6424–6431
Braggio E, Van Wier S, Ojha J, McPhail E, Asmann YW, Egan J et al (2015) Genome-wide analysis uncovers novel recurrent alterations in primary central nervous system lymphomas. Clin Cancer Res 21:3986–3994. https://doi.org/10.1158/1078-0432.Ccr-14-2116
Cameron DL, Jacobs N, Roepman P, Priestley P, Cuppen E, Papenfuss AT (2021) VIRUSBreakend: viral integration recognition using single breakends. Bioinformatics 37:3115–3119. https://doi.org/10.1093/bioinformatics/btab343
Cavaliere R, Petroni G, Lopes MB, Schiff D (2010) Primary central nervous system post-transplantation lymphoproliferative disorder: an International Primary Central Nervous System Lymphoma Collaborative Group Report. Cancer 116:863–870. https://doi.org/10.1002/cncr.24834
Chapman JR, Bouska AC, Zhang W, Alderuccio JP, Lossos IS, Rimsza LM et al (2021) EBV-positive HIV-associated diffuse large B cell lymphomas are characterized by JAK/STAT (STAT3) pathway mutations and unique clinicopathologic features. Br J Haematol 194:870–878. https://doi.org/10.1111/bjh.17708
Chapuy B, Roemer MG, Stewart C, Tan Y, Abo RP, Zhang L et al (2016) Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood 127:869–881. https://doi.org/10.1182/blood-2015-10-673236
Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA (2018) Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol Biol 1711:243–259. https://doi.org/10.1007/978-1-4939-7493-1_12
Chen H, Lee JM, Wang Y, Huang DP, Ambinder RF, Hayward SD (1999) The Epstein-Barr virus latency BamHI-Q promoter is positively regulated by STATs and Zta interference with JAK/STAT activation leads to loss of BamHI-Q promoter activity. Proc Natl Acad Sci USA 96:9339–9344. https://doi.org/10.1073/pnas.96.16.9339
Chen H, Lee JM, Zong Y, Borowitz M, Ng MH, Ambinder RF et al (2001) Linkage between STAT regulation and Epstein-Barr virus gene expression in tumors. J Virol 75:2929–2937. https://doi.org/10.1128/jvi.75.6.2929-2937.2001
Doubrovinskaia S, Sahm F, Thier MC, Bendszus M, Wick W, Seliger C et al (2021) Primary CNS lymphoma after CLIPPERS: a case series. J Neurol Neurosurg Psychiatry 92:1348–1349. https://doi.org/10.1136/jnnp-2020-325759
Fink SE, Gandhi MK, Nourse JP, Keane C, Jones K, Crooks P et al (2014) A comprehensive analysis of the cellular and EBV-specific microRNAome in primary CNS PTLD identifies different patterns among EBV-associated tumors. Am J Transplant 14:2577–2587. https://doi.org/10.1111/ajt.12858
Flieswasser T, Van den Eynde A, Van Audenaerde J, De Waele J, Lardon F, Riether C et al (2022) The CD70-CD27 axis in oncology: the new kids on the block. J Exp Clin Cancer Res 41:12. https://doi.org/10.1186/s13046-021-02215-y
Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J et al (2019) GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47:D766-d773. https://doi.org/10.1093/nar/gky955
Frigault MJ, Dietrich J, Gallagher K, Roschewski M, Jordan JT, Forst D et al (2022) Safety and efficacy of tisagenlecleucel in primary CNS lymphoma: a phase 1/2 clinical trial. Blood 139:2306–2315. https://doi.org/10.1182/blood.2021014738
Fukumura K, Kawazu M, Kojima S, Ueno T, Sai E, Soda M et al (2016) Genomic characterization of primary central nervous system lymphoma. Acta Neuropathol 131:865–875. https://doi.org/10.1007/s00401-016-1536-2
Gandhi MK, Hoang T, Law SC, Brosda S, O’Rourke K, Tobin JWD et al (2021) EBV-associated primary CNS lymphoma occurring after immunosuppression is a distinct immunobiological entity. Blood 137:1468–1477. https://doi.org/10.1182/blood.2020008520
Gebauer N, Künstner A, Ketzer J, Witte HM, Rausch T, Benes V et al (2021) Genomic insights into the pathogenesis of Epstein-Barr virus-associated diffuse large B-cell lymphoma by whole-genome and targeted amplicon sequencing. Blood Cancer J 11:102. https://doi.org/10.1038/s41408-021-00493-5
Giunco S, Celeghin A, Gianesin K, Dolcetti R, Indraccolo S, De Rossi A (2015) Cross talk between EBV and telomerase: the role of TERT and NOTCH2 in the switch of latent/lytic cycle of the virus. Cell Death Dis 6:e1774. https://doi.org/10.1038/cddis.2015.145
Grande BM, Gerhard DS, Jiang A, Griner NB, Abramson JS, Alexander TB et al (2019) Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 133:1313–1324. https://doi.org/10.1182/blood-2018-09-871418
Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O’Donnell E et al (2012) Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res 18:1611–1618. https://doi.org/10.1158/1078-0432.Ccr-11-1942
Gupta NK, Nolan A, Omuro A, Reid EG, Wang CC, Mannis G et al (2017) Long-term survival in AIDS-related primary central nervous system lymphoma. Neuro Oncol 19:99–108. https://doi.org/10.1093/neuonc/now155
Hadjadj J, Castro CN, Tusseau M, Stolzenberg M-C, Mazerolles F, Aladjidi N et al (2020) Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun 11:5341. https://doi.org/10.1038/s41467-020-18925-4
Haile WB, Gavegnano C, Tao S, Jiang Y, Schinazi RF, Tyor WR (2016) The Janus kinase inhibitor ruxolitinib reduces HIV replication in human macrophages and ameliorates HIV encephalitis in a murine model. Neurobiol Dis 92:137–143. https://doi.org/10.1016/j.nbd.2016.02.007
Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G et al (2004) Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 103:275–282. https://doi.org/10.1182/blood-2003-05-1545
Hernández-Verdin I, Kirasic E, Wienand K, Mokhtari K, Eimer S, Loiseau H et al (2023) Molecular and clinical diversity in primary central nervous system lymphoma. Ann Oncol 34:186–199. https://doi.org/10.1016/j.annonc.2022.11.002
Hoang-Xuan K, Deckert M, Ferreri AJM, Furtner J, Gallego Perez-Larraya J, Henriksson R et al (2023) European Association of Neuro-Oncology (EANO) guidelines for treatment of primary central nervous system lymphoma (PCNSL). Neuro Oncol 25:37–53. https://doi.org/10.1093/neuonc/noac196
Kaulen LD, Erson-Omay EZ, Henegariu O, Karschnia P, Huttner A, Günel M et al (2021) Exome sequencing identifies SLIT2 variants in primary CNS lymphoma. Br J Haematol 193:375–379. https://doi.org/10.1007/s11060-019-03208-w
Kaulen LD, Galluzzo D, Hui P, Barbiero F, Karschnia P, Huttner A et al (2019) Prognostic markers for immunodeficiency-associated primary central nervous system lymphoma. J Neurooncol 144:107–115. https://doi.org/10.1007/s11060-019-03208-w
Kaulen LD, Gumbinger C, Hinz F, Kessler T, Winkler F, Bendszus M et al (2022) Intraventricular immune checkpoint inhibition with nivolumab in relapsed primary central nervous system lymphoma. Neurooncol Adv 4:vdac051. https://doi.org/10.1093/noajnl/vdac051
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Li JSZ, Abbasi A, Kim DH, Lippman SM, Alexandrov LB, Cleveland DW (2023) Chromosomal fragile site breakage by EBV-encoded EBNA1 at clustered repeats. Nature. https://doi.org/10.1038/s41586-023-05923-x
Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E et al (2018) The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun 9:1558. https://doi.org/10.1038/s41467-018-04013-1
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D et al (2021) The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 23:1231–1251. https://doi.org/10.1093/neuonc/noab106
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Ma Y, Visser L, Roelofsen H, de Vries M, Diepstra A, van Imhoff G et al (2008) Proteomics analysis of Hodgkin lymphoma: identification of new players involved in the cross-talk between HRS cells and infiltrating lymphocytes. Blood 111:2339–2346. https://doi.org/10.1182/blood-2007-09-112128
Mangolini M, Maiques-Diaz A, Charalampopoulou S, Gerhard-Hartmann E, Bloehdorn J, Moore A et al (2022) Viral transduction of primary human lymphoma B cells reveals mechanisms of NOTCH-mediated immune escape. Nat Commun 13:6220. https://doi.org/10.1038/s41467-022-33739-2
McKenna M, Epperla N, Ghobadi A, Liu J, Lazaryan A, Ibrahim U et al (2023) Real-world evidence of the safety and survival with CD19 CAR-T cell therapy for relapsed/refractory solid organ transplant-related PTLD. Br J Haematol. https://doi.org/10.1111/bjh.18828
Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G (2011) GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12:R41. https://doi.org/10.1186/gb-2011-12-4-r41
Mottok A, Renné C, Seifert M, Oppermann E, Bechstein W, Hansmann M-L et al (2009) Inactivating SOCS1 mutations are caused by aberrant somatic hypermutation and restricted to a subset of B-cell lymphoma entities. Blood 114:4503–4506. https://doi.org/10.1182/blood-2009-06-225839
Motwani M, Pesiridis S, Fitzgerald KA (2019) DNA sensing by the cGAS–STING pathway in health and disease. Nat Rev Genet 20:657–674. https://doi.org/10.1038/s41576-019-0151-1
Moulignier A, Lamirel C, Picard H, Lebrette MG, Amiel C, Hamidi M et al (2017) Long-term AIDS-related PCNSL outcomes with HD-MTX and combined antiretroviral therapy. Neurology 89:796–804. https://doi.org/10.1212/wnl.0000000000004265
Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F et al (2019) Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37:773–782. https://doi.org/10.1038/s41587-019-0114-2
Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C et al (2022) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2015–2019. Neuro Oncol 24:v1–v95. https://doi.org/10.1093/neuonc/noac202
Radke J, Ishaque N, Koll R, Gu Z, Schumann E, Sieverling L et al (2022) The genomic and transcriptional landscape of primary central nervous system lymphoma. Nat Commun 13:2558. https://doi.org/10.1038/s41467-022-30050-y
Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A et al (2017) Genetic and functional drivers of diffuse large B cell lymphoma. Cell 171:481-494.e15. https://doi.org/10.1016/j.cell.2017.09.027
Reumers J, De Rijk P, Zhao H, Liekens A, Smeets D, Cleary J et al (2011) Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat Biotechnol 30:61–68. https://doi.org/10.1038/nbt.2053
Rossi D, Rasi S, Fabbri G, Spina V, Fangazio M, Forconi F et al (2012) Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 119:521–529. https://doi.org/10.1182/blood-2011-09-379966
Salaverria I, Martin-Guerrero I, Wagener R, Kreuz M, Kohler CW, Richter J et al (2014) A recurrent 11q aberration pattern characterizes a subset of MYC-negative high-grade B-cell lymphomas resembling Burkitt lymphoma. Blood 123:1187–1198. https://doi.org/10.1182/blood-2013-06-507996
Santosha V, Anas Y (2016) The immune microenvironment in Hodgkin lymphoma: T cells, B cells, and immune checkpoints. Haematologica 101:794–802. https://doi.org/10.3324/haematol.2015.132761
Sauer T, Parikh K, Sharma S, Omer B, Sedloev D, Chen Q et al (2021) CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood 138:318–330. https://doi.org/10.1182/blood.2020008221
Schaff LR, Grommes C (2022) Primary central nervous system lymphoma. Blood 140:971–979. https://doi.org/10.1182/blood.2020008377
Shanmugam V, Craig JW, Hilton LK, Nguyen MH, Rushton CK, Fahimdanesh K et al (2021) Notch activation is pervasive in SMZL and uncommon in DLBCL: implications for Notch signaling in B-cell tumors. Blood Adv 5:71–83. https://doi.org/10.1182/bloodadvances.2020002995
Sorrentino A, Menevse AN, Michels T, Volpin V, Durst FC, Sax J et al (2022) Salt-inducible kinase 3 protects tumor cells from cytotoxic T-cell attack by promoting TNF-induced NF-κB activation. J Immunother Cancer 10:e004258. https://doi.org/10.1136/jitc-2021-004258
Stiller CA, Chessells JM, Fitchett M (1994) Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer 70:969–972. https://doi.org/10.1038/bjc.1994.431
Talevich E, Shain AH, Botton T, Bastian BC (2016) CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 12:e1004873. https://doi.org/10.1371/journal.pcbi.1004873
Tang C-HA, Lee AC, Chang S, Xu Q, Shao A, Lo Y et al (2021) STING regulates BCR signaling in normal and malignant B cells. Cell Mol Immunol 18:1016–1031. https://doi.org/10.1038/s41423-020-00552-0
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P (2015) Sambamba: fast processing of NGS alignment formats. Bioinformatics 31:2032–2034. https://doi.org/10.1093/bioinformatics/btv098
Tesselaar K, Arens R, van Schijndel GMW, Baars PA, van der Valk MA, Borst J et al (2003) Lethal T cell immunodeficiency induced by chronic costimulation via CD27-CD70 interactions. Nat Immunol 4:49–54. https://doi.org/10.1038/ni869
The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium (2020) Pan-cancer analysis of whole genomes. Nature 578:82–93. https://doi.org/10.1038/s41586-020-1969-6
Tison A, Garaud S, Chiche L, Cornec D, Kostine M (2022) Immune-checkpoint inhibitor use in patients with cancer and pre-existing autoimmune diseases. Nat Rev Rheumatol 18:641–656. https://doi.org/10.1038/s41584-022-00841-0
Vater I, Montesinos-Rongen M, Schlesner M, Haake A, Purschke F, Sprute R et al (2015) The mutational pattern of primary lymphoma of the central nervous system determined by whole-exome sequencing. Leukemia 29:677–685. https://doi.org/10.1038/leu.2014.264
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF et al (2012) A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. New Engl J Med 366:799–807. https://doi.org/10.1056/NEJMoa1110557
Villarino AV, Kanno Y, O’Shea JJ (2017) Mechanisms and consequences of Jak–STAT signaling in the immune system. Nat Immunol 18:374–384. https://doi.org/10.1038/ni.3691
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164. https://doi.org/10.1093/nar/gkq603
Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C et al (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306:269–271. https://doi.org/10.1126/science.1102160
Weniger MA, Melzner I, Menz CK, Wegener S, Bucur AJ, Dorsch K et al (2006) Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation. Oncogene 25:2679–2684. https://doi.org/10.1038/sj.onc.1209151
Wood DE, Lu J, Langmead B (2019) Improved metagenomic analysis with Kraken 2. Genome Biol 20:257. https://doi.org/10.1186/s13059-019-1891-0
Zhu Y, An X, Zhang X, Qiao Y, Zheng T, Li X (2019) STING: a master regulator in the cancer-immunity cycle. Mol Cancer 18:152. https://doi.org/10.1186/s12943-019-1087-y
Acknowledgements
The authors would like to thank Dr. Nagarajan Paramasivam, Dr. Philip Reiner Kensche, and the DKFZ Omics IT and Data Management Core Facility (ODCF) for technical support with WES data processing. Dr. Leon D. Kaulen has been funded by a fellowship of the DKFZ Clinician Scientist Program, supported by the Dieter Morszeck Foundation.
Funding
Open Access funding enabled and organized by Projekt DEAL. No targeted funding reported.
Author information
Authors and Affiliations
Contributions
LDK, FH, FS, and WW recruited the cohort. LDK and FH performed retrospective chart review. FH and FS performed immunohistochemistry of specimen, provided pathological evaluation, and performed DNA and RNA extraction. LDK, ED, DF, LH, BB, FS and WW analyzed sequencing data. LDK, AK, PL, and JI performed validation experiments. LDK and ED wrote the first draft. All authors analyzed and interpreted data, critically revised, read and approved the final manuscript. LDK, FS and WW designed and oversaw this study.
Corresponding authors
Ethics declarations
Conflict of interest
Dr. Kaulen reports no disclosures. Dr. Denisova reports no disclosures. Dr. Hinz reports no disclosures. Ms. Hai reports no disclosures. Mr. Friedel reports no disclosures. Dr. Henegariu reports no disclosures. Mr. Hoffmann reports no disclosures. Mr. Ito reports no disclosures. Mr. Kourtesakis reports no disclosures. Mr. Lehnert reports no disclosures. Dr. Doubrovinskaia reports no disclosures. Dr. Karschnia reports no disclosures. Dr. von Baumgarten reports no disclosures. Dr. Kessler reports no disclosures. Dr. Baehring reports no disclosures. Dr. Brors reports no disclosures. Dr. Sahm reports honoraria from Illumina, Agilent and Bayer, patents on methylation-based classification managed by DKFZ, and being cofounder and shareholder of Heidelberg Epignostix GmbH. Dr. Wick reports support for clinical trials from Apogenix, Pfizer, Roche and Vaximm as well as honoraria for advice from Astra Zeneca, Enterome, MSD, Roche, Servier and Vaximm paid to the Medical Faculty.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaulen, L.D., Denisova, E., Hinz, F. et al. Integrated genetic analyses of immunodeficiency-associated Epstein-Barr virus- (EBV) positive primary CNS lymphomas. Acta Neuropathol 146, 499–514 (2023). https://doi.org/10.1007/s00401-023-02613-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-023-02613-w