
Abstract
Background:
The Paul-Ehrlich-Institut (PEI) published an analysis of reports of adverse drug reactions (ADRs) in 2001 with test and therapy allergens from the period from 1991 to 2000. Possible risk factors were evaluated for the ADR reports classified as “serious”. During the analyzed period, modified semi-depot preparations (allergoids) induced between 0.01 % and 0.0005 % serious systemic reactions, i. e. one serious ADR occurred in 10,000 to 200,000 injections. No information was provided regarding the respective incidences in relation to the individual companies or preparations.
Materials and methods:
Within the scope of a 13-year analysis (2001–2013), the serious ADRs were analysed at HAL for the allergoid preparations PURETHAL® Pollen and Mites. As in the analysis of PEI, the frequency of serious ADRs was based on the estimated number of administered injections.
Results:
A total of 46 cases with serious ADRs were received. In 26 % of the cases, a serious ADR occurred during initial treatment after the first injection. In 82.6 % of the cases, the serious ADR occurred within the 30-minute observation period in the practice. Adrenaline was administered as emergency treatment in seven cases. Hospitalisations were initiated by the treating physician or by patients themselves in 45 cases. The duration of the hospital stay varied from a few hours up to three days for further monitoring. Serious ADRs occurred in 11 cases with mites and in 35 cases with pollen. If it is assumed that there are six injections in each vial, this yields an incidence of 0.00061 % (1 : 164,000). The frequency with mites (0.00093 %) was slightly higher than with pollen (0.00055 %).
Conclusions:
The allergoid preparations (pollen and mite allergens) showed a very low risk of serious ADRs, which was close to the lower level of incidence of 0.0005 % for allergoid preparations published by the PEI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Specific subcutaneous immunotherapy (SCIT) is an established method of therapy for the treatment of allergic IgE-mediated diseases [1]. Semi-depot preparations were introduced by Fuchs and Strauss [2] in 1959. The release of the allergen is delayed by binding to a carrier substance (e. g. aluminium hydroxide) in these preparations. By introducing semi-depot preparations, tolerance has considerably improved [3]. Further development of native semi-depot allergen extracts by means of chemical modification (formaldehyde, glutaraldehyde) [4, 5, 6] to so-called allergoid preparations had the objective to further increase tolerance. In addition, initial treatment (increasing dose) could be reduced to a few injections in comparison with traditional multi-strength semi-depot preparations [7].
Further and continuous training of doctors and the practice team, within the scope of emergency seminars, has also possibly contributed to a reduction in serious adverse reactions [8, 9, 10]. Furthermore, in 1996 the manufacturers of allergen preparations were obliged by the Paul-Ehrlich-Institut (PEI) to incorporate a warning (“Hyposensitization vaccines for injection should only be prescribed and applied by doctors trained or experienced in allergy”) in the summary of product characteristics (SPC) as well as in the patient information [10, 11].
Systemic ADRs with fatal outcome have fortunately decreased, despite an increased number of patients treated and thus injections administered, as shown by the figures for the period from 1981 to 1988 and from 1991 to 2000 [9, 11].
In the PEI publication, only the ADR reports for test and therapy allergens classified as “serious” were assessed with regard to possible risk factors [11]. “Anaphylactic reactions were assessed as serious if they corresponded to stage III or IV…, were life-threatening, required emergency treatment or short-term inpatient monitoring or required a sickness certificate for a few days.”
In addition to this, a serious ADR is defined in the current version of the German Drug Law (Arzneimittelgesetz) as: “an adverse reaction that is fatal or life-threatening, requires inpatient hospitalisation or a prolongation of an inpatient hospitalisation, results in permanent damage or a severe impairment, disability or congenital anomalies or birth defects” [12]. In case one of these conditions is applicable, the ADR is classified as serious.
In total 1,100 events were reported, of which 555 were assessed as serious after subcutaneous injection [11]. A further long-term analysis carried out by PEI covering a later period is not available to date.
The S2 guideline (2009) of the allergy associations [13] contains the following information relating to tolerance data after 2001: “Data within the scope of approval extensions between 2001 and 2005 yielded no evidence of a changed frequency of serious ADRs compared with the figures published in 2001.”
Overall, in the PEI analysis, frequencies between 0.002 % and 0.0077 % were reported for unmodified semi-depot allergens in relation to the estimated number of administered injections and values between 0.0005 % and 0.01 % were observed for chemically modified semi-depot preparations [11, 14].
In the publication by Siefert [9] from 1990, which was published 11 years before, the estimated rate of adverse reactions was reported as 0.0022 % and 0.0087 %. “This means that in 100,000 injections, two to nine adverse reactions of corresponding severity have been omitted.”
The aim of the present analysis, is to assess how the incidence is depicted with regard to serious ADRs for the allergoid preparations PURETHAL Mites and PURETHAL Pollen compared with the PEI analysis from the year 2001 [11] and whether other aspects regarding safety could be found. In order to compare the results, the same approach was used as described in the publication of PEI.
Material and Methods
The ADRs spontaneously reported in Germany and classified as serious for the preparation PURETHAL® (HAL Allergy B.V., Leiden) with the allergens pollen and house dust mites (HDM) were analysed from the database (Saphëus 2009 PV Database, PHARMSOFT Dr. B. Rodust, Ascheberg) for the period stated. The assessment of an ADR as a “serious” adverse drug reaction was made analogously to the definition outlined in the PEI analysis [11].
“The assessment of the risk of this treatment can only be made based on a basic consideration of the relative frequencies of the serious systemic reactions which were reported during a defined period of time. In addition to the absolute number of reports, the knowledge of the quantities of the medication given during this period is also required. Here the PEI relies on safety reports and ‘postmarketing’ studies by the manufacturer” [11].
The sales figures for each year revealed the total amount of the medicinal product in the examined period. These numbers were used to calculate the frequencies.
The calculation of the frequencies has been done as described in the PEI Publication [11]: The number of packs sold and the estimated injections administered (six injections per vial in the maintenance treatment) were used as a basis for the calculation.
No differentiation was made according to the number of injections in the initial treatment (IT) and the number of injections in the maintenance treatment (MT). An estimation of six injections per vial was used for both IT and MT.
Analogous to the PEI publication, the reports were categorized according to age, gender, applied allergens, emergency treatment (use of adrenaline), duration of monitoring/treatment in hospital, initial or maintenance treatment, possible risk factors (in particular allergic bronchial asthma), immediate or late reactions, temporal occurrence of the ADR and allergen involved, quantitative distribution of the allergens and potential quality defects in the preparations used.
Results
During the assessment period from January 2001 to December 2013, 46 cases were reported where the patient experienced a serious adverse reaction (43 of these suffered a systemic reaction) after application of the allergoid preparation. In 45 cases the patient was hospitalized, in some cases only for a few hours for further monitoring. In a few cases, the patient presented at the hospital without referral after experiencing a late reaction.
Assessment according to gender, age and allergen prescribed
Distribution according to gender did not show a difference (24 females, 22 males). Sixteen children experienced serious ADR 8 adolescents and 22 adults (Tab. 1). The distribution of allergens in children, adolescents and adults yielded no “risk allergen” in the three age groups.
Emergency treatment and use of adrenaline
Adrenaline was used in seven cases. In one case emergency treatment was given, but not specified according to substance class. It therefore remained open whether this patient also received adrenaline.
Duration of the hospital stay and/or the treatment
For ten patients no information was reported. One patient from this group was not admitted to the hospital due to an immediate adverse reaction, but for the diagnostic investigation of another disease. Details on hospital duration were reported in 36 cases. In 21 cases, it was reported that they stayed in the hospital for a few hours, for one day or one night. In two cases from this group it was not clear whether the patient left the hospital on the same evening.
Twelve patients were admitted for 2 days and three patients for 3 days.
Hospitalisations by the doctor or visits to the outpatient department by the patient were documented in 45 cases in total after injection of SIT.
Occurrence of the ADR during initial treatment (IT) or maintenance treatment (MT)
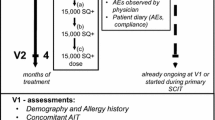
The assessment of the ADR reports with regard to the occurrence of reactions showed that in 31 (67.4 %) cases the serious ADR occurred during IT, of which in 12 (26.1 %) cases the ADR occurred after the first injection (see Fig 1).

The number of cases during initial treatment (IT) (from 0.05 ml to 0.5 ml) or in maintenance treatment (MT) (0.5 ml)
© Distler, A.
In 15 cases (32.6 %), the patient experienced a serious ADR after reaching the highest dose of 0.5 ml. The serious ADRs during MT occurred from between a few weeks of treatment until just before the end of treatment in the third year of SIT.
Patients receive six injections in 5 weeks during IT followed by further injections at delayed intervals (4 +/- 2 weeks) during MT. Therefore, the majority of injections are administered during MT. Fig. 1 shows that the frequency of a serious ADR during the IT is higher than during the subsequent three years of MT.
Possible risk factors, in particular allergic bronchial asthma
Several risk factors were identified which may have either in part triggered or caused the adverse reaction:
-
Allergic bronchial asthma: Three patients with a history of asthma experienced an asthma attack during SCIT; two in the IT and one in the MT.
-
A past history of psychological illnesses (e. g. anxiety neuroses) was reported by doctors in two cases.
-
Phobia of needles: One patient (22 years old) with known phobia of needles developed circulatory collapse five minutes after the first injection (0.05 ml).
-
Hypertension: A person suffering from hypertension (43 years old) receiving no adequate treatment with concomitant risk factors (stress and high workload) developed hypertension.
-
Several concomitant illnesses: One patient (41 years old) with hypertension, obesity and asthma as a concomitant illness reacted systemically during IT.
-
Increase in dose during the pollen season: One patient (23 years old) with an allergy to trees and grass pollen, developed an ADR after the injection of 0.3 ml from a mixture of tree and grass, while the patient already suffered from slight allergic symptoms during the tree pollen season.
In the 46 cases, there was no evidence of an intravasal allergen influx, overdose or exceeding the treatment interval. It could not be determined whether an additional allergen strain, severe sensitisation, a latent infection or current psychological strain played a role here.
Allergic asthma was reported in twelve cases (26.1 %) as a concomitant illness on the ADR form, but of these patients only three patients responded with an asthma attack during SIT. In one case, the patient (12 years old) had already suffered from an asthma attack on the day of the injection, although she did not inform the treating doctor of this. Seven minutes after the injection of 0.1 ml, another asthma attack occurred, which then led to hospitalisation.
In eight other cases, an ADR with involvement of the lungs (dyspnoea, coughing, inspiratory stridor) after the injection was reported. In total, eleven patients (23.9 %) experienced a serious ADR involving the lungs.
Immediate reactions (< 30 minutes after the injection) and late reactions (> 30 minutes)
In 29 cases (63 %), the ADR occurred within the 30-minute observation period. In nine cases no time of onset was reported, however, in these cases emergency treatment had been initiated in the practice, which suggests start of the ADR within 30 minutes. Immediate reactions therefore probably occurred within the observation period in 38 cases (82.6 %).
Seven (15.2 %) late reactions (>30 minutes after injection) were also documented by the reporting doctors. One patient with a delayed ADR had received two allergen extracts on the same day.
Temporal occurrence of the ADR and allergens involved
There was a temporal relationship between the occurrence of the reaction and the allergens used in terms of the late reactions. The seven late reactions all occurred with pollen preparations. Nevertheless, due to the low number of cases reported (n = 46), this observation is not very reliable.
Quantitative allergen distribution
In 35 cases pollen allergens triggered the occurrence of the AE and in 11 cases, HDM (Fig. 2).

Distribution of the allergens (pollen and mites), whose injection resulted in serious adverse drug reactions after injection.
© Distler, A.
Taken together, pollen is therefore cited as the cause for hospitalisation more often than HDM; however, in relation to the number of packages sold the latter is more often the causative allergen for serious ADRs.
Quality deficits in the preparations used as the cause for a serious ADR
This was not stated or suspected in any case. With the exception of mugwort, the pollen preparations were all approved by the PEI. Both pollen and HDM (since 2008, when the Therapy Allergen Ordinance [Therapieallergene-Verordnung] in Germany became obligatory) are both subjected to batch testing and release by PEI.
Calculation of the frequency of serious ADRs
A frequency of 0,00061 % (based on an estimation of six injections per vial) was defined for these allergoid preparations.
This means that within 164,000 injections one serious ADR (mostly with hospital admission) occurred.
If only the pollen allergens are taken into consideration, serious ADRs occured in 0.00055 % (1 : 182,000) of the injections. The frequency of HDM was slightly higher than that with pollen at 0.0009 % (1 : 107,500).
Overall, an allergic or anaphylactic reaction (n = 26 patients) was most commonly the reason for hospitalisation (Tab. 2). In three patients asthma, in six cases cardiovascular symptoms and in three cases a massive local reaction was the reason for hospital admission. Eight patients were referred to hospital for monitoring or further treatment due to other “allergic or immunological reactions“.
In the majority of cases, the hospital stay served only for monitoring after the emergency treatment of the patient in the practice.
Discussion
Allergy emergencies can have various causes. SIT with allergen extracts can even lead to anaphylactic reactions. The treating doctor must be able to treat anaphylaxis and must try to identify possible risk factors prior to the injection and avoid these in future [15].
In this publication, the attempt was made to ascertain possible risk factors for serious reactions in SIT, which are, in part, also listed in the guidelines of the allergy associations for SIT [13].
The reported 46 cases show, that by more careful medical history prior to injection, or by compliance with the SPC (e. g. no dose increase during the pollen season, treat only asthmatics with expiratory volume in 1 second [FEV1] > 70 %) the onset of a serious AE could have been prevented. The statement that the assessment of ADR reports within the scope of spontaneous recording does not draw conclusions in terms of the frequency of occurrence of the ADR [11] is very true. In the context of spontaneous reporting, the analysis of ADR reports cannot give any conclusion on the frequency of ADRs.
In contrast to clinical studies, in which every adverse reaction is carefully documented, in a spontaneous recording system, the doctor’s willingness to document information is an important factor. Local and mild systemic ADRs are rarely reported by doctors (to date). Diverse information is available in the literature regarding ADRs depending on allergens, preparation and dose schedule [16, 17, 18, 19].
It is assumed that many adverse reactions, in particular those with mild symptoms, are not reported by doctors resulting in reporting bias. It can be assumed that this reporting bias is lower for serious ADRs, which are usually accompanied by hospitalisation. The documentation will probably be more thorough and the reporting frequency higher, than in the case of non-serious adverse reactions. According to an investigation, a small amount of doctors reported ADRs in the past.
According to estimates, even serious ADRs are only recorded in approximately 10 % of cases [20], however, in the field of allergy, the reporting ratio may be higher. Therefore it cannot be ruled out that “underreporting” leads to inaccurate results and incorrect conclusions. In addition, it cannot be ruled out that the risk of serious ADRs is actually higher in reality than calculated in this assessment.
In 2007, the Drug Commission of the German Medical Association (Arzneimittelkommission der deutschen Ärzteschaft) published guidelines regarding reporting ADRs in children [21]. How Adverse Events (AE) can be reported quickly and easily, was published in the German journal “Ärzteblatt” [22].
The reasons for “underreporting” are multifactorial, where the time required and the lack of remuneration are often reported by doctors.
The number of reports in the PEI publication [11] which contained 555 serious ADRs — divided into two five-year analyses each (1991–1995 and 1996–2000) — was much higher than the number of serious cases reported to HAL for the period 2001–2013.
According to the investigation by the PEI [11], 88 % of the 555 serious reactions occurred within 30 minutes.
Of the 46 cases assessed here, the ADR occurred within the 30-minute observation period following the injection in 38 patients (82.6 %). This result therefore roughly corresponds with that of the PEI.
However, in another study with a five-year monitoring period assessing the systemic ADRs (2005–2010), the authors reported that only 52 % of the ADRs occurred within the 30-minute observation period [23]. It was not stated whether the patients were hospitalised. However, 60 % of the patients received emergency treatment including amongst others adrenalin.
The search for risk factors in the PEI publication [11], found that physical or psychological stress factors could not be ruled out in around 3 % for the period from 1991 to 1995 and around 8 % for the period from 1996 to 2000.
In the 46 cases analyzed here, according to the patient history five (10.9 %) suffered from physical or mental conditions.
The PEI assessed asthma as a warning sign for developing a serious ADR [11]. In 238 patients who experienced a serious ADR during the period from 1995 to 2000, 79 suffered from mild to moderate asthma and two patients had severe asthma (34 % of patients in total).
In this evaluation, allergic asthma as a concomitant disease had been observed by the reporting physician in 26 % cases. Three patients (6.5 %) suffered from an asthma attack and were directed to the hospital for follow-up. This number of cases is too small to conclude that allergic asthma is a risk signal. In 24 % of AE reports a condition involving the lungs was reported by physician was documented.
In the analysis of PEI the allergen causing the serious ADRs could be identified in 460 out of the 555 ADR. The ADR was caused in 37 % by an allergen mixture of grass and cereals, in 17 % by an allergen mixture of grasses, trees and herb pollen, in 15 % by a mite mixture, in 14 % by tree pollen and in 12 % by grass pollen.
Of the 46 cases evaluated in this publication, in around 4 % of the cases the ADR was caused by grasses plus cereals, in 33 % by grasses, in 15 % by tree pollen, in 24 % by mixtures of grasses and trees and in 24 % by mites. According to both analyses, grasses and the mixtures of grasses plus cereal pollen are the allergen groups with the most reported cases. Without exact sales figures for the individual allergens, no conclusion can be made on possible “risk allergens”.
Grasses or grasses plus cereal pollen were definitely the most frequently prescribed allergen in the past and the number of reported ADRs is not surprising.
The risk-benefit assessment evaluated in the PEI publication [11] can be used to verify whether the safety profile of the PURETHAL preparations is similar to the frequency range specified for the allergoids by PEI.
The PEI calculated frequencies of 0.01 % (1 : 12,500) to 0.0055 % (1 : 200,000) [14] for chemically modified preparations (allergoids).
In the analysis of the 46 cases received during this, even longer, period (13 years instead of 10 years), the frequency rate of 0.00061 % (1 : 164,000) per injection was observed.
In order to calculate the risk for a defined period of time, the number of cases and the amount of medication dispensed are required.
These three key figures were available and have been used for the calculation of risk of serious ADRs in accordance with the procedure of the PEI. In both reporting periods, 10 or 13 years, the factor which should be taken into account is the willingness to report by physicians.
The assessment carried out by the PEI — separated into two time periods — showed a reduction in the number of reported ADRs during the second recording period (1996 to 2000), whilst at the same time the reports of serious ADRs significantly increased. During the second five-year period, there was a significant increase in the severe asthma reactions in patients with a history of known obstructive bronchitis or asthma.
However, a separate calculation of the frequencies for both periods was not performed by the PEI.
An evaluation of the cases reported as serious for the period from 2001 to 2010 [24] only yielded 24 reports. In 2011 to 2013, a further 22 cases were reported, which are included in this evaluation. The reasons for this increase are multifactorial, in our opinion. The increased administration numbers resulting in an increase in the number of patients treated could be the reason for this. “More intensive” training of the sales representatives in pharmacovigilance and a greater willingness to report by doctors could also play a role here. It is also possible that more patients with contraindications, such as pulmonary diseases, received SCIT in recent years.
The frequency was calculated as 0.00046 % for the period from 2001 to 2010 (10 years) and was therefore lower than the calculation three years (2001 to 2013) later (for 13 years) at 0.00061 %. This shows that the calculation of the risk of serious ADRs for each injection results in different values for the analysed period selected. It seems useful to carry out an analysis over a longer time period, as was done by the PEI [9, 10, 11] in the past. A further calculation of frequency for the period after 2001 is not available.
In the S2 guideline of allergy societies [13] from the year 2009, the statement is found that there has been no evidence of an altered frequency of serious ADR reports with respect to the figures published in 2001.
Compared to the analysis period of the PEI publication, an increase in serious cases could be determined in the last three years.
When examining the serious cases divided according to allergens (pollen vs. mites), a slightly higher rate of cases was observed for mite preparations compared with pollen preparations. The number of reported cases with mites (n = 11 of 46 patients) was too small to evaluate the possible causes.
When examining the age distribution it showed that 24 of the 46 cases were children and adolescents. There was no evidence of an increased frequency in this age group. The tolerance data of a one-year application study and the subgroup analysis results of 339 children and adolescents did not show any differences in the tolerance between children and adult patients [25]. There is no information regarding the age distribution of the patients in the PEI publication [11]. Since the absolute number of treated children and adolescents in the period 2001 to 2013 was not available, no conclusions on the patient’s age and the risk of ADRs can be made. When considering the distribution of the cases between IT and MT, the following should be taken into consideration: Since the calculation was based on the vials sold during the period stated and as this concerned an allergen preparation with only one vial, it cannot be determined how many injections were administered during IT and during MT (with 0.5 ml per injection). The distribution of the cases during IT and the number during MT are shown in Fig. 1.
As all patients receive at first the IT and this is followed by the MT, the analysis of the reported data might give the impression that more serious AE occur in the IT than during the MT.
The aim of this assessment was not to make statements on a patient basis. Patients can receive either preseasonal or perennial treatment with this allergen preparation. The interval between the injections may be between four (+/-2) weeks in MT and may be administered differently by doctors than is recommended in the SPC. Therefore, the number of injections a patient actually receives in the recommended three-year treatment period depends on different aspects. In the PEI publication, however, it was established that the assessment of the case reports with regard to the occurrence of reactions during the initial or maintenance phase showed an increase in the adverse reactions during MT.
After analysing the 46 cases, the first injection resulted in a serious ADR in 12 (26.1 %) cases. This shows that SCIT vaccinations for injection should be administered and used by doctors who are trained in allergy or by experienced allergists.
On the contrary, recent studies with an even faster up-dosing and omission of the first dose of 0.05 ml, show a good tolerance in adults (“Rush”- schedule with three injections) [26] and in children with mite allergies (“cluster” - schedule with two injections in one day) [27]. This faster up-dosing with three injections has been approved for the pollen allergoid preparation for adults with allergies to grass pollen since 2011.
The guideline published in 2007 “Acute emergency treatment of anaphylactic reactions”, and the revised version published in 2014, advises early use of adrenaline in the case of anaphylactic reactions [28]. Adrenaline was used only in seven of the 46 cases during the above period.
Conclusion
The allergoid preparation discussed in this publication has a low risk of causing serious ADRs (with hospitalisation) in relation to the number of packs sold and estimated number of injections administered. The determined frequency of 0.00061 % for the period 2001 to 2013 was within the lower range of the frequency of 0.01–0.0005 % published by the PEI in 2001 for allergoid preparations (period from 1991 to 2000).
Abbreviations
- MT:
-
Maintenance treatment
- FEV1:
-
Forced expiratory volume in one second
- IT:
-
Initial treatment
- HDM:
-
House dust mites
- PEI:
-
Paul-Ehrlich-Institut
- SCIT:
-
Specific subcutaneous immunotherapy
- SIT:
-
Specific immunotherapy
- ADR:
-
Adverse drug reactions
- SPC:
-
Summary of product characteristics
References
Noon L. Prophylactic inoculation against hay fever. Lancet 1911;1:1572–1573
Fuchs AM, Strauss MB. The clinical evaluation and the preparation and standardization of suspension of a new water-insoluble whole ragweed pollen complex. J Allergy 1959;30:66–82
Bousquet J, Lockey RF, Malling HJ. Allergen immunotherapy: therapeutic vaccines for allergic diseases. A WHO Position Paper. Allergy 1998;53:1–42
Maasch HJ, Marsch DG. Standardized extracts modified allergens-allergoids. Clin Rev Allergy 1987;5:89–106
Marsh DG, Norman PS, Roebber M, Lichtenstein LM. Studies on allergoids from naturally occurring allergens. III. Preparation of ragweed pollen allergoids by aldehyde modification in two steps. J Allergy Clin Immunol 1981;68:449–59
Akdis CA, Blaser K. Mechanisms of allergen-specific immunotherapy. Allergy 2000;55:522–30
Zimmermann T, Beinder K, Mollet FE, Kroon AM. Therapie von Heuschnupfen und Pollenasthma bei Kindern mit einem neuen, modifizierten Allergenextrakt. Immunität und Infektion 1990;18:127–31
Haffner HT. Letale Komplikationen bei Desensibilisierungsbehandlungen: Aspekte zur Untersuchung und rechtsmedizinischen Begutachtung. Zentralbl Rechtsmed 1985;27:765–73
Siefert G. Nebenwirkungen bei der Hyposensibilisierung, Ursachenanalyse - Zukunftsperspektiven. Allergologie 1990;13:150–5
Lüderitz-Püchel, U, May S, Haustein D. Zwischenfälle nach Hyposensibilisierung. Münch Med Wochenschr 1996;138:129–48
Lüderitz-Püchel U, Keller-Stanislawski B, Haustein D. Neubewertung des Risikos von Test- und Therapieallergenen. Eine Analyse der UAW Meldungen von 1991 bis 2000. Bundesgesundheitsbl Gesundheitsforsch Gesundheitsschutz 2001;44:709–18
Arzneimittelgesetz (AMG) i.d.F. d. Bek. v. 12.12.2005, BGBI. I S.3394; §4, Abs. 13
Kleine-Tebbe J, Bergmann KC, Friedrichs F, Fuchs T, Jung K, Klimek L et al. Die spezifische Immuntherapie (Hyposensibilisierung) bei IgE-vermittelten allergischen Erkrankungen. Allergo J 2006;15:56–74
Lüderitz-Püchel U. Risikobewertung von Test- und Therapie-Allergenen. Eine Analyse der UAW-Meldungen von Januar 1991 bis Dezember 2000. In: Jorde W, Mönchengladbacher Allergie-Seminar Bd. 14. München-Deisenhofen: Dustri; 2003. p. 19–28
Bauer C, Ruëff F, Przybilla B. Notfälle in der Allergologie. Hautarzt 2004;55:525–32
Iglesias-Cadarso A, Hernández-Weigand P, Reaño M, Pérez-Pimiento, Vargas Núñez JA, Torre F de la. Risk factors for systemic reactions to allergen-specific subcutaneous immunotherapy. J Investig Allergol Clin Immunol 2010;20:621–2
Mellerup MT, Hahn GW, Poulsen LK, Malling H. Safety of allergen-specific immunotherapy. Relation between dosage regimen, allergen extract, disease and systemic side-effects during induction treatment. Clin Exp Allergy 2000;30:1423–9
Amin HS, Liss GM, Bernstein DI. Evaluation of near-fatal reactions to allergen immunotherapy injections. J Allergy Clin Immunol 2006;117:169–75
Gastaminza G, Algorta J, Audicana M, Etxenagusia M, Fernandez E, Munoz D. Systemic reactions to immunotherapy: influence of composition and manufacturer. Clin Exp Allergy 2003;33:470–4
Göttler M, Munter KH, Hasford J et al. Zu viele Ärzte sind „meldemüde“. Dtsch Arztebl 1999;96:1221-3
Arzneimittelkommission der deutschen Ärzteschaft, ed. Leitfaden zur Meldung unerwünschter Arzneimittelwirkung bei Kindern. Dtsch Arztebl 2007;104:A 1533–34
Gundert-Remy U, Stammschulte T. Unerwünschte Arzneimittelwirkungen. Jetzt einfach und schnell melden. Dtsch Arztebl 2010;107:A 1434
Melamed J, Mehra A, Ahuja-Malim A. A 5-year study of systemic reactions using both shared and patient-specific vaccines. Allergy Rhinol 2013;4:e88–e93
Distler A, Pafferath B. 10-Jahres-Übersicht zu unerwünschten Arzneimittelwirkungen (UAW) mit Klinikeinweisung nach einer spezifischen Immuntherapie (SIT) mit einem chemisch modifizierten Allergenpräparat (Allergoid). Allergo J 2011;20:41
Distler A, Meijerman A, Kleinjans H. Safety and clinical efficacy of an allergoid depot preparation in children and adolescents in daily practice. Allergy 2010;65:574
Pfaar O, Twuijer E van, Hecker H, Boot JD, Ree R van, Klimek L. Accelerated up-dosing of subcutaneous immunotherapy with a registered allergoid grass pollen preparation. Int Arch Allergy Immunol 2013;160:420–4
Peñas A, Carcía-González M, Cruz MJ, Valdesoiro L, Boot JD, Larramona H et al. Observational study of the safety of a cluster schedule for subcutaneous immunotherapy in a pediatric population. J Investig Allergol Clin Immunol 2013;23:63–5
Ring J, Beyer K, Biedermann T, Bircher A, Duda D, Fischer J et al. Guideline for acute therapy and management of anaphylaxis. S2 guideline of DGAKI, AedA, GPA, DAAU, BVKJ, ÖGAI, SGAI, DGAI, DGP, DGPM, AGATE and DAAB. Allergo J Int 2014;23:96–112
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of interest
Debbie Pappelendam is an employee of HAL Allergy B.V. in Leiden, the Netherlands.
Dr. Andreas Distler is an employee of HAL Allergie GmbH in Düsseldorf, Germany.
Cite this as
Distler A, Pappelendam D. 13-year overview of severe adverse drug reactions following subcutaneous specific immunotherapy with a chemically modified allergen preparation. Allergo J Int 2015;24:294–302 DOI: 10.1007/s40629-015-0083-z
Rights and permissions
About this article

Cite this article
Distler, A., Pappelendam, D. 13-year overview of serious adverse drug reactions following subcutaneous specific immunotherapy with a chemically modified allergen preparation. Allergo J Int 24, 294–302 (2015). https://doi.org/10.1007/s40629-015-0084-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40629-015-0084-y