
Abstract
Heteroplasmy, the presence of two or more variants in an organism, may render mitochondrial DNA (mtDNA)-based individual identification challenging in forensic analysis. However, the variation of heteroplasmic proportions and the segregation of heteroplasmic variants through generations and within families have not been systematically described at a large scale in animals such as the domestic dog. Therefore, we performed the largest study to date in domestic dogs and screened a 582-bp-long fragment of the mtDNA control region in 180 individuals in 58 pedigrees for signs of heteroplasmy. We identified three pedigrees (5.17%) with heteroplasmic point mutations. To follow the segregation of the point mutations, we then analyzed 131 samples from these three independent pedigrees and found significant differences in heteroplasmy between generations and among siblings. Frequently (10% of cases), the proportion of one base changed from 0–10% to 80–90% (as judged from Sanger electropherograms) between generations and varied to a similar extent among siblings. We included also a literature review of heteroplasmic and potential mutational hot spot positions in the studied region which showed that all heteroplasmic positions appear to be mutational hot spots. Thus, although heteroplasmy may be used to increase the significance of a match in forensic case work, it may also cause erroneous exclusion of related individuals because of sharp switches from one state to the other within a single generation or among siblings especially in the presented mutational hot spots.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondrial DNA (mtDNA) is a genetic marker frequently used in forensic analysis [1 and references therein], and in population genetic and phylogenetic research [2, 3 and references therein]. The advantages of this marker are manifold. A high copy number in cells enables analysis of small amounts of cell material (e.g., [1]) which is especially important in forensic studies where samples often contain small amounts and/or highly degraded DNA. Because of a high mutation rate, compared to nuclear DNA, analysis of a relatively short mtDNA region is often sufficient for forensic [1] identification or fine-scale phylogeographical studies [2, 3]. In general, uniparental (maternal) inheritance as well as lack of recombination of mtDNA simplifies the reconstruction of pedigree analysis and population history [2, 3].
However, heteroplasmy [4, 5] may render analysis in forensic case work difficult. Heteroplasmic organisms carry more than one mitochondrial genotype (e.g., [1, 6, 7]). Three possible mechanisms may generate heteroplasmy: paternal leakage, recombination, and mutation [2, 3 and references therein]. Paternal leakage and recombination seem to be rare events normally not leading to heteroplasmy; instead mutations in the haploid mitochondrial genome are the most important cause. Heteroplasmic point mutations clearly present a challenge for correct individual identification [1, 6] in forensics and for haplotype identification in population genetic/phylogenetic studies [2, 3].
As a consequence of mutation, heteroplasmy represents the intermediate step to fixation of either the wild type or the mutated type [5, 8, 9] and is therefore an essential step in the generation of genetic diversity in mtDNA. For a neutral mitochondrial heteroplasmic variant, the time to fixation has been estimated to be approximately 200 generations in humans and chinook salmon [2]. However, there have been cases reporting extremely fast fixation of heteroplasmic point mutations, within a few generations (e.g., [10, 11]) and in an extreme case in one generation [12]. The rapid segregation of haplotypes is thought to be generated by a bottleneck during oogenesis or embryogenesis reducing the number of mtDNA molecules, leading to random genetic drift [13–15]. Therefore, the proportions of heteroplasmic components may vary among siblings or from mother to offspring [16, 17]; e.g., one sibling may have an apparently fixed, homoplasmic genotype while others display extensive variation of proportions. In forensic case work, this may lead to related individuals incorrectly being ruled as nonrelated. Variable levels of heteroplasmy may even occur within individuals between tissues (mosaicism) [1, 18–21] and may therefore lead to incorrect haplotype assignment and exclusion of an individual as source of an evidence material. On the other hand, occurrence of the same heteroplasmic point mutation in otherwise identical sequences was used to increase the significance of a match for identifying the Romanov family members [22–24]. Forensic guidelines for analysis of human samples allow for one sequence difference between individuals without excluding the possibility that the individuals belong to the same lineage (e.g., [25]). However, for dogs, the majority of haplotypes, based on the control region, differ by a single mutation from each other [26]. Therefore, basing exclusions on at least one sequence differences may not be practical since match probabilities would often be too high for an informative analysis. The reason for this low degree of difference between haplotypes is that dogs were domesticated only 10,000–15,000 years ago [26] and, consequently, few sequence differences have accumulated since the origin of dogs.
In dogs, the control region of the mtDNA genome has been used to study the geographical origin and age of dogs [26–28] as well as their migration routes, e.g., of the Dingo to Australia or the Old World origin of New World dogs [29, 30]. In addition, the mtDNA control region has been widely used for the identification of dogs in forensic analyses [31–39]. It has been proposed to standardize the nomenclature of the mtDNA control region in order to launch a dog database for forensic use [40] and complementary information about heteroplasmic states and mutational hot spots may be useful in this context. All of the aforementioned utilizations warrant the systematic study and identification of heteroplasmy in the mtDNA control region in dogs.
Although heteroplasmy has been occasionally reported in dogs [20, 35, 41], to our knowledge, there are no extensive studies of this phenomenon in dogs. Also, in other animal species, very few studies [11, 42] have evaluated how heteroplasmy levels vary among related individuals and between generations (but see, e.g., [8, 43] for studies in humans). This is probably because samples from far-related specimens or family/pedigree information are not available in most animal species. The domestic dog with extensively recorded pedigrees (e.g., the Swedish and Finnish Kennel Club databases), dense populations, and short generation times provides a unique opportunity to study heteroplasmy on a larger scale. In this study, we perform the first comprehensive study of heteroplasmy in a large number of dog pedigrees. We identify pedigrees with heteroplasmic point mutations and study the segregation and variation of heteroplasmy between generations and among siblings as well as between distant parts of the pedigrees. We also study the correlation between nucleotide positions showing heteroplasmy and mutational hot spots, and we discuss the implications of these results for forensic studies.
Materials and methods
Samples
Pedigree information was obtained from the public databases available through the Swedish (http://kennet.skk.se/hunddata/) and Finnish (http://jalostus.kennelliitto.fi) Kennel Clubs (Supplementary Tables 1 and 2). The representation of the samples analyzed in this study is shown in pedigrees (Fig. 1a–c; Supplementary Figs. 1, 2, and 3). Great care was taken to collect samples from different parts of the pedigree and to include several dogs from the same litters. The samples were collected as buccal epithelial cells using FTA-indicating cards according to the manufacturer’s specifications (Whatman International, UK) or as EDTA–blood samples from which genomic DNA was extracted using a commercially available kit (Puregene; Gentra Systems, Minneapolis, MN, USA). In order to test whether the level of heteroplasmy differs between tissues, we tested 18 dogs for both buccal cells and blood samples. As an additional quality measure, eight blood samples and 12 buccal samples were sequenced twice to compare the possible differences between reruns of the same sample.

Simplified pedigrees A, B, and C. Squares males, circles females, black filled squares/circles sampled males/females. Vertical lines present one generation; the number of horizontal lines crossing vertical lines indicates additional generations. The dashed line in pedigree B indicates that the sample Z1766 belongs to this pedigree, but the exact number of generations is unknown. The dashed box in pedigree C indicates that only in this part of the pedigree heteroplasmic sequences are found. Numbers 1–10 refer to the litters given in Table 1
mtDNA sequence analysis
Amplification and sequencing of a 582-bp-long fragment of the mtDNA control region was performed as described before [34]. Four primer pairs were used to score each heteroplasmic position three times and in both forward and reverse directions. Sequencing of PCR products was performed with BigDye Terminator chemistry on ABI 377 and ABI 3700 instruments (Applied Biosystems). Sequences were aligned with BioEdit [44] and checked by eye. Comparisons of sequences and identification of haplotypes were performed with DnaSP v5 [45].
Sequences and contigs were visually checked to guarantee optimal sequence quality with a minimum of background signal. Proportions of major and minor component were scored by eye (Supplementary Table 2). The proportion of heteroplasmy was estimated by assessing the height of the two peaks at the heteroplasmic position and calculating the proportion between the heights of each peak respective to the sum of the height of both peaks (similar to [8]). Importantly, direct sequencing approaches have been found to be able to score minor component levels of ~10% [9, 43]. Therefore, what appears as a fixed state may be a heteroplasmic state with the minor component at a frequency of less than 10%.
The proportions of the three respective electropherograms were averaged. For the indel case, the peak height of the visible nucleotide was compared to the nearest nucleotides because the deletion was obviously not possible to score. This was feasible because neighboring nucleotides generally showed congruent peak height differences relative to the heteroplasmic position in all three electropherograms. However, it is worth pointing out that these measures represent approximations because sequence quality may differ due to signal intensity and nucleotide incorporation and lab conditions [9 and references therein].
Comparison of heteroplasmic proportions between runs of the same dog sample
In order to ensure that laboratory conditions were stable and to assess the difference between runs, we separately amplified and sequenced eight blood samples and 12 buccal cell samples twice. For blood samples, five samples were scored with identical proportions in both runs and three differed by 10% between runs. For buccal cell samples, seven samples showed identical proportions in both runs and the other five differed by 5–10% between runs (Supplementary Table 2). All samples showed the same predominant nucleotide and were therefore identified as the same haplotype.
Comparison of heteroplasmic proportions between blood and buccal cell samples from the same dog
The difference in heteroplasmic proportions between buccal cell and blood samples from the same dog were assessed for a subset of nine heteroplasmic individuals. One sample had identical scores in buccal cell and blood sample, but the other eight cases showed discrepancies: by a maximum of 10% in heteroplasmic proportions in four cases, 20% in one case, 35% in two, and by a maximum of 40% in one case (Supplementary Table 2). In conclusion, in all cases, heteroplasmy could be identified when amplifying from different DNA sources, but the proportion of heteroplasmy differed more between different DNA sources than between different runs from the same DNA source. Thus, in general, there was good agreement between runs and the few cases which showed higher discrepancies did not impact our analysis noticeably. However, where possible, only one source (either buccal cell or saliva sample) was used for closely related individuals in order to avoid potential bias in heteroplasmic scores.
Results
Screening of pedigrees for heteroplasmies
We studied altogether 180 dogs in 58 pedigrees for signs of heteroplasmy by sequencing a 582-bp-long fragment of the mtDNA control region (nps 15,458–16,039). We used two to 12 dogs from each pedigree corresponding to a total minimum of 875 generations (3,469 years; Supplementary Table 1). Interestingly, the average generation time was 3.96 years (based on the birth year of sampled dogs, and going back in time to the most recent common ancestor of each pedigree), which is in good accordance to previous estimates for canids [46]. Out of the 58 pedigrees, we found three pedigrees (5.17%) with heteroplasmic point mutations, two samples from each pedigree having different haplotypes. The nucleotide positions were clearly heteroplasmic when checked by eye. It should be noted that the resolution of the used approach is limited (see “Materials and methods” section) and cannot detect low levels of heteroplasmy (<10%), and it is possible that some heteroplasmic pedigrees may have been missed.
Segregation of heteroplasmies through generations
To identify the characteristics of heteroplasmy, we studied additional dogs in the three identified pedigrees with heteroplasmy. A total of 131 dogs were studied and are subsequently referred to as pedigrees A, B, and C (Fig. 1; Supplementary Figs. 1, 2, and 3). The studied samples were chosen from litters and across generations to cover the entire pedigrees. We found heteroplasmy at nucleotide positions (pos) 16,003 (G/A), 15,639 (G/A), and 15,931 (A/−). All three positions have been found to vary between haplotypes within three or four out of the six major phylogenetic clades A-F (pos 16,003, clades B–D; pos 15,639, A–D; pos 15,931, A–D) previously identified from dogs [26–28]. Therefore, it appears that these positions mutate more frequently than other positions.
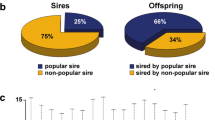
For the smallest pedigree, B (Fig. 1b; Supplementary Fig. 2), 11 samples were analyzed, spanning a minimum of 107 years and 32 generations (average generation time 3.34 years). Out of these, seven samples had a heteroplasmic point mutation at position 16,003 resulting in a heteroplasmic mix of G/A (G and A giving haplotypes D1 and D4, respectively [47]). The other four samples carried the mutated haplotype D4. D1 and D4 belong to a haplogroup called d1, and it has been shown that haplogroup d1 is almost exclusively found in Scandinavia in spitz-type dog breeds [47] and therefore this special haplogroup may be especially informative in forensic case work. Taking also this specific heteroplasmy into account, a dog may be assigned with high confidence to pedigree B (and to Lapponian herders) because, so far, only dogs belonging to this pedigree show this heteroplasmy.
For pedigree C (Fig. 1c; Supplementary Fig. 3), we analyzed 46 dogs representing 120 generations (627 years) with an average generation time of 5.23 years. The pedigree had a heteroplasmic point mutation at pos 15,639 with a heteroplasmic mix of G/A (G and A giving haplotypes B1 and B35, respectively). Notably, this lineage showed heteroplasmic dogs (seven samples) only in one lineage (indicated by a black-framed box in Fig. 1c and Supplementary Fig. 3) out of five descending from the founder individual, and all samples from other parts were homoplasmic for the wild-type nucleotide G (38 samples).
Remarkably, a second heteroplasmy was identified in the same part of pedigree C (black-framed box in Fig. 1c and Supplementary Fig. 3). One dog (Z2178) which was homoplasmic wild-type G at pos 15,639 (giving haplotype B1) had a heteroplasmic single base indel at another position [15,931; the same position as the heteroplasmy in pedigree A (see below)] resulting in a mix of A/− (A and − giving the haplotypes B1 and B3, respectively). Thus, each heteroplasmy occurred independently in different dogs. A second sample (Z2225), which is closely related to Z2178 (Fig. 1c; Supplementary Fig. 3), showed apparently the wild-type nucleotide at both pos 15,639 and pos 15,931. Both dogs were sampled twice at independent occasions, and each sample was run twice to confirm sequence data. Thus, in this part of the pedigree, close relatives carried two different heteroplasmies or were homoplasmic for the wild type, potentially resulting in three different haplotype scores (B1, B3, and B35).
Finally, pedigree A (Fig. 1a; Supplementary Fig. 1) had a heteroplasmic single base indel at pos 15,931 with mixes of A/− (A and − giving haplotypes D1 and D3, respectively). Heteroplasmy occurred in all parts of the pedigree and 26 samples were heteroplasmic, 36 samples had the wild-type nucleotide A, whereas 12 samples had an apparent fixation to the deletion (−100%). The 74 samples analyzed across the pedigree encompassed 137 generations and 533 years, giving an average generation time of 3.89 years.
Proportions of heteroplasmy at different kinship levels
Within heteroplasmic parts of the pedigrees, proportions of the respective nucleotides varied considerably among dogs between apparent homoplasmy for each haplotype and any proportions in between (Fig. 1, Table 1). Proportions of heteroplasmy differed strikingly at all kinship levels, demonstrated best by pedigree A (Fig. 1; Supplementary Fig. 1). At the sibling level (within the same litter), some siblings had identical or nearly identical proportions of heteroplasmic point mutations while some had 100% of one of the two nucleotides and therefore looked like having fixed haplotypes. For example, among ten litters with at least three members, the heteroplasmic proportions differed by 100% in one litter (i.e., each opposite fixed state was carried by two siblings) and by 80% in two other litters (Table 1). The detection limit in Sanger sequencing for a minority peak is approximately 10%, and a proportion up to 20% may easily be overlooked. Therefore, in at least 10% of cases, siblings would normally have been scored with different haplotypes.
Between generations, 30 mother–offspring pairs could be studied (Fig. 2). In general, the whole range of possible nucleotide proportion differences was observed. Thirteen cases (43.3%) included nucleotide proportion changes of 60% or more, thereby leading to changes in the haplotype score. Most importantly, three out of 30 mother–offspring pairs (10%) had proportion differences of 80–90% between generations. Thus, at the 10% detection level of minority peaks, switches between two apparently fixed states appeared frequently. Accordingly, at larger distances in the pedigrees, the probability of an erroneous exclusion of a dog is even higher. For example, dog Y418 and the sibling pair Y426 and Y427 (pedigree A; Fig. 1) are cousins (their mothers are siblings). Also here a total shift with apparent fixation of two different haplotypes has occurred which would lead to exclusion of kinship at the cousin level in dogs. Further, closely related litters in pedigree C (black framed box, Fig. 1c) showed full fixation of the wild-type allele in one litter (Z2147 and Z2141), whereas the other litter (litter 10 in Table 1; Z2137, Z2027, and Z2163) showed a heteroplasmic mix of wild-type (40%) and mutated allele (60%) with the mutated allele in majority. Further, shifts of heteroplasmic proportions among closely related dogs (e.g., Z2139, Z2146, and Z2135) covering three to four generations could be observed. Thus, the heteroplasmic proportions differ strongly and randomly between generations, and the pattern of transmission suggests that severe bottlenecks are involved in mtDNA inheritance in dogs. Comparing different main branches of the pedigrees, no clear pattern could be identified except that pedigree C showed heteroplasmy in only one branch of the pedigree. In the other pedigrees, all parts of the pedigrees showed full range of heteroplasmic proportions and therefore heteroplasmy persists through several generations in these pedigrees.

Difference of heteroplasmic proportions among mother–offspring pairs in percent
Discussion
We present here a comprehensive study of heteroplasmy in canine pedigrees. We show that close relatives or even siblings cannot be reliably used for forensic identification because the proportions of wild-type (WT) and mutant (MT) nucleotide vary considerably among siblings and across generations. Close relatives may show total reversion of WT and MT proportions which may lead to an erroneous exclusion of case material. Additionally, the special case of two independent mutations and therefore three different haplotypes in one of the studied pedigrees further challenge case studies in dogs.
Intergenerational changes have been previously shown in a series of other animals and in human (e.g., [8, 10–12, 43]). For example, [43] found that five out 135 human mother–child comparisons had a strong shift in heteroplasmic point mutations (two point heteroplasmies found in mother and resolved to homoplasmy in children; three point heteroplasmies found in children only). The full fixation of the mutant variant within one or two generations in the North Atlantic right whale, Eubalaena glacialis, resulting in different haplotypes between siblings, has been identified by McLeod and White [11]. Finally, Santos et al. [8] studied 48 human pedigrees (422 individuals) corresponding to 321 mtDNA transmissions and identified both shifts (up to ~40% change in heteroplasmic proportions) in some pedigrees as well as an apparently stable pattern of heteroplasmy in other pedigrees. In the present study, it could be confirmed that rapid switches of heteroplasmic point mutations occur between generations among dogs and that different offspring may carry opposite genotypes (of the wild-type or mutant variant) or show a range of different heteroplasmic proportions.
It has been proposed that heteroplasmy may occur more frequently at hypervariable sites (mutational hot spots) [6, 9, 48–50]. All three heteroplasmic positions identified in this study have shown variation in previous studies and are possibly mutational hot spots. To study the connection between heteroplasmy and hypervariability, we compiled data from this study and from literature about these two phenomena for positions within the 582-bp region (Table 2). The positions were identified based on having shown heteroplasmy and/or signs of hypervariability [26, 41].
-
1.
A study of 105 dogs from the UK by Wetton et al. [20] found a heteroplasmic single base indel in one dog in a blood sample. The heteroplasmic proportions ranged from near fixation of the deletion to predominance of the wild-type nucleotide in a sample of 12 hairs from the same dog. This heteroplasmy (15,931; A/−) occurred at the same position as in pedigree A.
-
2.
Five point heteroplasmic positions among 133 dogs from Austria, four of which within the region sequenced in the present study, were detected by Eichmann and Parson [35] by direct sequencing. One of these positions (15,931; A/−) was the same as in pedigree A and the case found by [20]. However, [35] found this position to be heteroplasmic in sequences from phylogenetic clades A and B and [20] in clade B, whereas we found it to be heteroplasmic in sequences from clade D in pedigree A (and clade B in pedigree C). Additionally, three other positions in HVS-I were found to vary considerably among clades and therefore suggested to be mutational hot spots (Table 2).
-
3.
A phylogenetic tree approach to calculate how often character changes (substitutions) have occurred at different nucleotide positions in the tree was applied by Gundry et al. [41]. They found three highly variable positions, with three to six substitutions in the phylogeny, in the 582-bp region studied in the present study. One of these, pos 15,639 with six changes and the three nucleotide states A/T/G, is identical to one of the two heteroplasmic positions in pedigree C in the present study.
-
4.
A recent study of 1,543 dogs by Pang et al. [26] increased the number of reported haplotypes considerably to 220 for the here studied 582-bp region (for comparison—[41] reported 45 haplotypes). Therefore, we checked if additional highly variable positions can be identified on the basis of the existing phylogeny with six major phylogenetic clades (A–F). Thus, mutated positions were identified by studying the alignments within each clade: more than one nucleotide, or an indel, at a position implies a mutation within a clade. Positions with at least three mutational changes were scored as a potential mutational hot spot. Position 15,639 carried a “C” in haplotype A165 and therefore all four character states (T/G/A/C) have now been observed in this position. Position 15,931 carried a “G” in haplotype A94 and in haplotypes found by [41] and therefore, in addition to the indel, also has an A–G substitution (A/G/−).
To conclude, all three heteroplasmic positions identified in the present study appear to be mutational hot spots. The indel position 15,931 which was heteroplasmic both in two of our pedigrees and in the studies by [20] and [35], and varies within phylogenetic clades A–D, has to be considered a mutational hot spot. The same applies to position 15,639 which changed six times in the phylogenetic tree of [41], carries all four possible nucleotides [26], and was heteroplasmic in our analysis. Finally, position 16,003 carries two different nucleotides in different haplotypes in clades B–D and therefore likely mutated at least three times in history and showed a heteroplasmic mix of both nucleotides in the current study. Thus, all three heteroplasmies identified in the present study are found at positions which are among the most variable. In total, only six positions have been reported so far to show heteroplasmy, and all these positions show mutations. This clearly demonstrates that heteroplasmy occurs more often at mutational hot spots.
In this study, we show that extreme shifts of heteroplasmic proportions can be observed among dogs both in mother–offspring pairs, and among siblings as well as other relatives, with, e.g., one dog (sibling or mother) carrying the wild type in apparently fixed state and siblings or offspring having the mutated type in fixation. Therefore, using heteroplasmy for increasing match significance of case material should be done with caution in dogs. The issue of how to quantify the match significance in these cases is also not yet solved. Further, the heteroplasmies occurred in what appears to be mutational hot spot positions with higher mutation rates than other nucleotide positions. It was also demonstrated that up to three haplotypes occurred in one part of a pedigree because mutational hot spots generated increased diversity. Thereby, it seems that mutational hot spot positions are unreliable for exclusions or confirmations of case material while other positions with lower mutation rates are more reliable. For example, possibly an exclusion based on differences in two hot spot mutations may be unreliable while a single difference occurring in a position with a low mutation rate may be highly informative. This fact may be taken into account in forensic analysis. A problem is that knowledge about which positions are hypervariable is largely missing and relies on studies of phylogenies and a few reported cases of heteroplasmy as reviewed in this study. The detailed pedigree analysis presented here contributes a first step to increased knowledge about the prevalence and nature of heteroplasmy and hotspots in dogs, but further detailed analyses are warranted.
References
Budowle B, Allard MW, Wilson MR, Chakraborty R (2003) Forensics and mitochondrial DNA: applications, debates, and foundations. Annu Rev Genomics Hum Genet 4:119–141. doi:10.1146/annurev.genom.4.070802.110352
White DJ, Wolff JN, Pierson M, Gemmell NJ (2008) Revealing the hidden complexities of mtDNA inheritance. Mol Ecol 17:4925–4942. doi:10.1111/j.1365-294X.2008.03982.x
Galtier N, Nabholz B, Glémin S, Hurst GDD (2009) Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol Ecol 18:4541–4550. doi:10.1111/j.1365-294X.2009.04380.x
Bendall KE, Macaulay VA, Baker JR, Sykes BC (1996) Heteroplasmic point mutations in the human mtDNA control region. Am J Hum Genet 59:1276–1287
Santos C, Sierra B, Álvarez L, Ramos A, Fernández E, Nogués R, Aluja MP (2008) Frequency and pattern of heteroplasmy in the control region of human mitochondrial DNA. J Mol Evol 67:191–200. doi:10.1007/s00239-008-9138-9
Melton T (2004) Mitochondrial DNA heteroplasmy. Forensic Sci Rev 16:1
Kmiec B, Woloszynska M, Janska H (2006) Heteroplasmy as a common state of mitochondrial genetic information in plants and animals. Curr Genet 50:149–159. doi:10.1007/s00294-006-0082-1
Santos C, Montiel R, Sierra B, Bettencourt C, Fernandez E, Alvarez L, Lima M, Abade A, Aluja MP (2005) Understanding differences between phylogenetic and pedigree-derived mtDNA mutation rate: a model using families from the Azores Islands (Portugal). Mol Biol Evol 22(6):1490–1505. doi:10.1093/molbev/msi141
Irwin JA, Saunier JL, Niederstätter H, Strouss KM, Sturk KA, Diegoli TM, Brandstätter A, Parson W, Parsons TJ (2009) Investigation of heteroplasmy in the human mitochondrial DNA control region: a synthesis of observations from more than 5000 global population samples. J Mol Evol 68:516–527. doi:10.1007/s00239-009-9227-4
Ashley MV, Laipis PJ, Hauswirth WW (1989) Rapid segregation of heteroplasmic bovine mitochondria. Nucl Acids Res 17:7325–7331
McLeod BA, White BN (2010) Tracking mtDNA heteroplasmy through multiple generations in the north Atlantic right whale (Eubalaena glacialis). J Hered 101(2):235–239. doi:10.1093/jhered/esp098
Koehler CM, Lindberg GL, Brown DR, Beitz DC, Freeman AE, Mayfield JE, Myers AM (1991) Replacement of bovine mitochondrial DNA by a sequence variant within one generation. Genetics 129:247–255
Jenuth JP, Peterson AC, Fu K, Shoubridge EA (1996) Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet 14:146–151
Cao L, Shitara H, Horii T, Nagao Y, Imai H, Abe K, Hara T, Hayashi J-I, Yonekawa H (2007) The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat Genet 39(3):386–390. doi:10.1038/ng1970
Cree LM, Samuels DC, de Sousa C, Lopes S, Rajasimha HK, Wonnapinij P, Mann JR, Dahl HHM, Chinnery PF (2008) A reduction in the number of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet 40:249–254. doi:10.1038/ng.2007.63
Lutz S, Weisser HJ, Heizmann J, Pollak S (1999) Mitochondrial heteroplasmy among maternally related individuals. Int J Leg Med 113:155–161. doi:10.1007/s004140050288
Sekiguchi K, Kasai K, Levin BC (2003) Inter- and intragenerational transmission of a human mitochondrial DNA heteroplasmy among 13 maternally-related individuals and differences between and within tissues in two family members. Mitochondrion 2:401–414. doi:10.1016/S1567-7249(03)00028-X
Bendall KE, Macaulay VA, Sykes BC (1997) Variable levels of a heteroplasmic point mutation in individual hair roots. Am J Hum Genet 61:1303–1308
Savolainen P, Arvestad L, Lundeberg J (2000) A novel method for forensic DNA investigations: repeat-type sequence analysis of tandemly repeated mtDNA in domestic dogs. J Forensic Sci 45:990–999
Wetton JH, Higgs JE, Spriggs AC, Roney CA, Tsang CSF, Foster AP (2003) Mitochondrial profiling of dog hairs. Forensic Sci Int 133:235–241. doi:10.1016/S0379-0738(03)00076-8
Paneto GG, Martins JA, Longo LVG, Pereira GA, Freschi A, Alvarenga VLS, Chen B, Oliveira RN, Hirata MH, Cicarelli RMB (2007) Heteroplasmy in hair: differences among hair and blood from the same individuals are still a matter of debate. Forensic Sci Int 173(2-3):117–121. doi:10.1016/j.forsciint.2007.02.011
Ivanov PL, Wadhams MJ, Roby RK, Holland MM, Weedn VW, Parsons TJ (1996) Mitochondrial DNA sequence heteroplasmy in the grand duke of Russia Georgij Romanov establishes the authenticity of the remains of Tsar Nicholas II. Nat Genet 12:417–420. doi:10.1038/ng0496-417
Coble MD, Loreille OM, Wadhams MJ, Edson SM, Maynard K, Meyer CE, Niederstätter H, Berger C, Berger B, Falsetti AB, Gill P, Parson W, Finelli LN (2009) Mystery solved: the identification of the two missing Romanov children using DNA analysis. PLoS ONE 4(3):e4838. doi:10.1371/journal.pone.0004838
Rogaev EI, Grigorenko AP, Moliaka YK, Faskhutdinova G, Goltsov A, Lahti A, Hildebrandt C, Kittler ELW, Morozova I (2009) Genomic identification in the historical case of the Nicholas II royal family. Proc Natl Acad Sci U S A 106(13):5258–5263. doi:10.1073/pnas.0811190106
SWGDAM (2003) Guidelines for mitochondrial DNA (mtDNA) nucleotide sequence interpretation. Forensic Sci Commun 5(2)
Pang JF, Kluetsch C, Zou XJ, Ab Z, Luo LY, Angleby H, Ardalan A, Ekström C, Sköllermo A, Lundeberg J, Matsumura S, Leitner T, Zhang YP, Savolainen P (2009) mtDNA data indicate a single origin for dogs south of Yangtze river, less than 16,300 years ago, from numerous wolves. Mol Biol Evol 26:2849–2864. doi:10.1093/molbev/msp195
Vilà C, Savolainen P, Maldonado JE, Amorim IR, Rice JE, Honeycutt RL, Crandall KA, Lundeberg J, Wayne RK (1997) Multiple and ancient origins of the domestic dog. Science 276:1687–1689. doi:10.1126/science.276.5319.1687
Savolainen P, Zhang YP, Luo J, Lundeberg J, Leitner T (2002) Genetic evidence of an East Asian origin of dogs. Science 298:1610–1613. doi:10.1126/science.1073906
Savolainen P, Leitner T, Wilton AN, Matisoo-Smith E, Lundeberg J (2004) A detailed picture of the origin of the Australian dingo, obtained from the study of mitochondrial DNA. Proc Natl Acad Sci U S A 101:12387–12390. doi:10.1073/pnas.0401814101
Leonard JA, Wayne RK, Wheeler J, Valadez R, Guillén S, Vilà C (2002) Ancient DNA evidence for Old World origin of New World dogs. Science 298:1613–1616. doi:10.1126/science.1076980
Savolainen P, Lundeberg J (1999) Forensic evidence based on mtDNA from dog and wolf hairs. J Forensic Sci 44:77–81
Schneider PM, Seo Y, Rittner C (1999) Forensic mtDNA hair analysis excludes a dog from having caused a traffic accident. Int J Leg Med 112:315–316. doi:10.1007/s004140050257
Aaspõllu A, Kelve M (2003) The first criminal case in Estonia with dog’s DNA data admitted as evidence. Int Congr Ser 1239:847–851. doi:10.1016/S0531-5131(02)00224-8
Angleby H, Savolainen P (2005) Forensic informativity of domestic dog mtDNA control region sequences. Forensic Sci Int 154(2–3):99–110. doi:10.1016/j.forsciint.2004.09.132
Eichmann C, Parson W (2007) Molecular characterization of the canine mitochondrial DNA control region for forensic applications. Int J Leg Med 121:411–416. doi:10.1007/s00414-006-0143-5
Hassell R, Heath P, Musgrave-Brown E, Ballard D, Harrison C, Thacker C, Catchpole B, Syndercombe Court D (2008) Mitochondrial DNA analysis of domestic dogs in the UK. Forensic Sci Int Genet Suppl Ser 1:598–599. doi:10.1016/j.fsigss.2007.10.187
Himmelberger AL, Spear TF, Satkoski JA, George DA, Garnica WT, Malladi VS, Smith DG, Webb KM, Allard MW, Kanthaswamy S (2008) Forensic utility of the mitochondrial hypervariable region 1 of domestic dogs, in conjunction with breed and geographic information. J Forensic Sci 53:81–89. doi:10.1111/j.1556-4029.2007.00615.x
Desmyter S, Comblez S (2009) Belgian dog mitochondrial DNA database for forensics. Forensic Sci Int Genet Suppl Ser 2(1):286–287. doi:10.1016/j.fsigss.2009.08.110
van Asch B, Albarran C, Alonso A, Angulo R, Alves C, Betancor E, Catanesi CI, Corach D, Crespillo M, Doutremepuich C, Estonba A, Fernandes AT, Fernandez E, Garcia AM, Garcia MA, Gilardi P, Gonçalves R, Hernández A, Lima G, Nascimento E, de Pancorbo MM, Parra D, de Fátima PM, Prat E, Puente J, Ramírez JL, Rendo F, Rey I, Di Rocco F, Rodríguez A, Sala A, Salla J, Sanchez JJ, Solá D, Silva S, Pestano Brito JJ, Amorim A (2009) Forensic analysis of dog (Canis lupus familiaris) mitochondrial DNA sequences: an inter-laboratory study of the GEP–ISFG working group. Forensic Sci Int Genet 4:49–54. doi:10.1016/j.fsigen.2009.04.008
Pereira L, van Asch B, Amorim A (2004) Standardisation of nomenclature for dog mtDNA D-loop: a prerequisite for launching a Canis familiaris database. Forensic Sci Int 141:99–108. doi:10.1016/j.forsciint.2003.12.014
Gundry RL, Allard MW, Moretti TR, Honeycutt RL, Wilson MR, Monson KL, Foran DR (2007) Mitochondrial DNA analysis of the domestic dog: control region variation within and among breeds. J Forensic Sci 52(3):562–572. doi:10.1111/j.1556-4029.2007.00425.x
Maté ML, Di Rocco F, Zambelli A, Vidal-Rioja L (2007) Mitochondrial heteroplasmy in control region DNA of South American camelids. Small Rumin Res 71:123–129. doi:10.1016/j.smallrumres.2006.04.016
Brandstätter A, Niederstätter H, Parson W (2004) Monitoring the inheritance of heteroplasmy by computer-assisted detection of mixed basecalls in the entire human mitochondrial DNA control region. Int J Leg Med 118:47–54. doi:10.1007/s00414-003-0418-z
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinforma 25:1451–1452. doi:10.1093/bioinformatics/btp187
Fuller KF, Mech LD, Cochrane JF (2003) Wolf population dynamics. In: Mech LD, Boitani L (eds) Wolves: behavior, ecology, and conservation. University of Chicago Press, Chicago
Klütsch CFC, Seppälä EH, Fall T, Uhlén M, Hedhammar Å, Lohi H, Savolainen P (2010) Regional occurrence, high frequency but low diversity of mitochondrial DNA haplogroup d1 suggests a recent dog–wolf hybridization in Scandinavia. Anim Genet. doi:10.1111/j.1365-2052.2010.02069.x
Stoneking M (2000) Hypervariable sites in the mtDNA control region are mutational hotspots. Am J Hum Genet 67:1029–1032
Melton T, Dimick G, Higgins B, Lindstrom L, Nelson K (2005) Forensic mitochondrial DNA analysis of 691 casework hairs. J Forensic Sci 50:73–80
Galtier N, Enard D, Radondy Y, Bazin E, Belkhir K (2006) Mutation hot spots in mammalian mitochondrial DNA. Genome Res 16:215–222. doi:10.1101/gr.4305906
Savolainen P, Arvestad L, Lundeberg J (2000) mtDNA tandem repeats in domestic dogs and wolves: mutation mechanism studied by analysis of the sequence of imperfect repeats. Mol Biol Evol 17:474–488
Acknowledgments
The authors would like to express their gratitude to numerous dog owners for their help in collecting samples and especially Iris Aintila for extensive sampling in Northern Sweden/Finland. We are thankful to Minna Virta and Sini Karjalainen for collection of samples and DNA isolation from blood samples. Further, we are very thankful to the Swedish and Finnish Kennel Clubs for providing essential information on their websites. This study was supported by the Academy of Finland, the Sigrid Juselius Foundation, Biocentrum Helsinki, the Jane and Aatos Erkko Foundation and the University of Helsinki Research Funds and by grants from the Swedish Research Council, and the Swedish Kennel Club. Peter Savolainen is a Royal Swedish Academy of Sciences Research Fellow supported by a grant from the Knut and Alice Wallenberg Foundation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Sampling list for screening 180 dogs from 58 lineages. Sample information including sample ID, haplotype name plus heteroplasmic proportions in the three pedigrees identified to carry heteroplasmic positions in the control region, geographical information, breed name, registration number of sampled dog, sex, name of sampled dog, name of father, name and registration number of mother, and other maternal ancestors back in time are given. N.B.: yellow-shaded boxes indicate the most recent common ancestors of groups of samples from the same pedigree, but also earlier maternal ancestors (name and registration number) are shown as a service for the community (XLS 123 kb)
Supplementary Table 2
Sampling list for screening 131 samples in the pedigrees identified to have a heteroplasmic position in the control region. Sample information include sample ID, geographical information, breed name, registration number, sex, and scored haplotype plus heteroplasmic proportions in the three pedigrees identified to carry heteroplasmic positions in the control region. Additionally, name of father and mother is given as well as all female ancestors (representing generations) for pedigree A (Pieta), pedigree B (Iresa), pedigree C (Söpö). *These samples were run twice in order to check for stable lab conditions. Minus sign indicates deletion. Additionally, siblings are highlighted in orange. Additional FTA card samples for comparison to blood samples are highlighted in turquoise. Note that three samples gave different haplotype scores when run for blood (Y) and buccal cell (Z) samples (Y438 and Z2195, Y439 and Z2204, and Y315/Z2162) (XLS 82 kb)
Supplementary Fig. 1
Detailed information for pedigree A. Squares males, circles females, black filled squares/circles sampled males/females. Red filled circles female ancestors (PDF 549 kb)
Supplementary Fig. 2
Detailed information for pedigree B. Squares males, circles females, black filled squares/circles sampled males/females. Red filled circles female ancestors. Dashed line indicates that the female “Loitsu” is a half-sister to I. Diidii, I. Davvenasti, and I. Darfi, but the number of generations is unknown (PDF 340 kb)
Supplementary Fig. 3
Detailed information for pedigree C. Squares males, circles females, black filled squares/circles sampled males/females. Red filled circles female ancestors. Black framed box indicates part of pedigree, in which both heteroplasmies are found (PDF 683 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Klütsch, C.F.C., Seppälä, E.H., Uhlén, M. et al. Segregation of point mutation heteroplasmy in the control region of dog mtDNA studied systematically in deep generation pedigrees. Int J Legal Med 125, 527–535 (2011). https://doi.org/10.1007/s00414-010-0524-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-010-0524-7