
Abstract
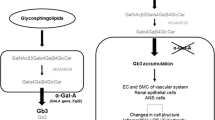
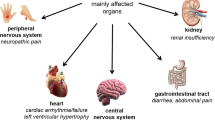
Fabry disease, the second most prevalent lysosomal storage disorder after Gaucher disease, is caused by mutations of the gene encoding the lysosomal hydrolase, α-galactosidase A. The enzymatic defect is inherited in an X-linked recessive fashion and leads to systemic glycosphingolipid deposition, resulting in profound dysfunction of neurological, renal, cardiac, and cerebrovascular systems. Although symptoms typically appear in childhood in hemizygous males and some heterozygous females, the diagnosis is often delayed or unrecognized, owing to variable presentations and low incidence. The initial phase begins in childhood or adolescence and is characterized by neuropathic pain, angiokeratomas, and ocular deposits. The later phase is distinguished by progressive cardiac, cerebral, and renal involvement, leading to multi-organ dysfunction and death. Recently published clinical trials have demonstrated the efficacy of enzyme replacement therapy in decreasing neuropathic pain and substrate deposition in target organs. Pediatricians have a key role to play in making the diagnosis, so that therapy can be initiated before irreversible tissue injury develops. Further research is required to determine optimal dosing protocols for treatment and to establish whether therapy can retard the progression of organ dysfunction, or even prevent these complications altogether.



Similar content being viewed by others

References
Brady RO, Schiffmann R (2000) Clinical features of and recent advances in therapy for Fabry disease. JAMA 284:2771–2775
Sessa A, Toson A, Nebuloni M, Pallotti F, Giordano F, Battini G, Maglio A, Meroni M, Calconi G, Bertolone G, Gatti P (2002) Renal ultrastructural findings in Anderson-Fabry disease. J Nephrol 15:109–112
Sweeley CC, Klionsky BL (1963) Fabry’s disease: classification as a sphingolipidosis and partial characterization of a novel glycolipid. J Biol Chem 238:3148–3150
Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L (1967) Enzymatic defect in Fabry’s disease: ceramide trihexosidase deficiency. N Engl J Med 276:1163–1167
Brady R, Grabowski G, Thadhani R (2001) Fabry disease: review and new perspectives. SynerMed Commun:1–8
Branton M, Schiffmann R, Kopp JB (2002) Natural history and treatment of renal involvement in Fabry disease. J Am Soc Nephrol 13 [Suppl 2]:S139–S143
Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, Grabowski G, Packman S, Wilcox WR (2003) Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med 138:338–346
Pastores GM, Thadhani R (2002) Advances in the management of Anderson-Fabry disease: enzyme replacement therapy. Expert Opin Biol Ther 2:325–333
Siamopoulos KC (2004) Fabry disease: kidney involvement and enzyme replacement therapy. Kidney Int 65:744–753
Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, Goldfarb L, Brady RO, Balow JE, Austin III HA, Kopp JB (2002) Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore) 81:122–138
Desnick R, Ioannou Y, Eng C (2001) Alpha-galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Kinzler KE, Vogelstein B (eds) The metabolic and molecular bases of inherited disease, 8th edn. McGraw Hill, New York, p 3733
MacDermot KD, Holmes A, Miners AH (2001) Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet 38:750–760
Alroy J, Sabnis S, Kopp JB (2002) Renal pathology in Fabry disease. J Am Soc Nephrol 13 [Suppl 2]:S134–S138
Meikle PJ, Hopwood JJ, Clague AE, Carey WF (1999) Prevalence of lysosomal storage disorders. JAMA 281:249–254
Nakao S, Kodama C, Takenaka T, Tanaka A, Yasumoto Y, Yoshida A, Kanzaki T, Enriquez AL, Eng CM, Tanaka H, Tei C, Desnick R (2003) Fabry disease: detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int 64:801–807
Ries M, Ramaswami U, Parini R, Lindblad B, Whybra C, Willers I, Gal A, Beck M (2003) The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr 162:767–772
Scott LJ, Griffin JW, Luciano C, Barton NW, Banerjee T, Crawford T, McArthur JC, Tournay A, Schiffmann R (1999) Quantitative analysis of epidermal innervation in Fabry disease. Neurology 52:1249–1254
Rowe JW, Gilliam JI, Warthin TA (1974) Intestinal manifestations of Fabry’s disease. Ann Intern Med 81:628–631
O’Brien BD, Shnitka TK, McDougall R, Walker K, Costopoulos L, Lentle B, Anholt L, Freeman H, Thomson AB (1982) Pathophysiologic and ultrastructural basis for intestinal symptoms in Fabry’s disease. Gastroenterology 82:957–962
Breunig F, Knoll A, Wanner C (2003) Enzyme replacement therapy in Fabry disease: clinical implications. Curr Opin Nephrol Hypertens 12:491–495
Mendez MF, Stanley TM, Medel NM, Li Z, Tedesco DT (1997) The vascular dementia of Fabry’s disease. Dement Geriatr Cogn Disord 8:252–257
DeGraba T, Azhar S, Dignat-George F, Brown E, Boutiere B, Altarescu G, McCarron R, Schiffmann R (2000) Profile of endothelial and leukocyte activation in Fabry patients. Ann Neurol 47:229–233
Frustaci A, Chimenti C, Ricci R, Natale L, Russo MA, Pieroni M, Eng CM, Desnick RJ (2001) Improvement in cardiac function in the cardiac variant of Fabry’s disease with galactose-infusion therapy. N Engl J Med 345:25–32
Scheidt W von, Eng C, Fitzmaurice T, Erdmann E, Hubner G, Olsen E, Christomanou H, Kandolf R, Bishop D, Desnick R (1991) An atypical variant of Fabry’s disease with manifestations confined to the myocardium. N Engl J Med 324:395–399
Meroni M, Sessa A, Battini G, Tazzari S, Torri Tarelli L (1997) Kidney involvement in Anderson-Fabry disease. Contrib Nephrol 122:178–184
Chen HC, Tsai JH, Lai YH, Guh JY (1990) Renal changes in heterozygous Fabry’s disease—a family study. Am J Kidney Dis 15:180–183
Farge D, Nadler S, Wolfe LS, Barre P, Jothy S (1985) Diagnostic value of kidney biopsy in heterozygous Fabry’s disease. Arch Pathol Lab Med 109:85–88
Okuda S (2000) Renal involvement in Fabry’s disease. Intern Med 39:601–602
Rodriguez FH Jr, Hoffmann EO, Ordinario AT Jr, Baliga M (1985) Fabry’s disease in a heterozygous woman. Arch Pathol Lab Med 109:89–91
Gubler MC, Lenoir G, Grunfeld JP, Ulmann A, Droz D, Habib R (1978) Early renal changes in hemizygous and heterozygous patients with Fabry’s disease. Kidney Int 13:223–235
Kriz W, Lemley K (1999) The role of the podocyte in glomerulosclerosis. Curr Opin Nephrol Hypertens 8:489–497
Clarke J GR, Wolfe L, Beaudoin J, Morehouse D (1972) Enzyme replacement therapy by renal allotransplantation in Fabry’s disease. N Engl J Med 287:1215–1218
Sessa A, Meroni M, Battini G, Maglio A, Nebuloni M, Tosoni A, Panichi V, Bertagnolio B (2002) Renal transplantation in patients with Fabry disease. Nephron 91:348–351
Maizel SE, Simmons RL, Kjellstrand C, Fryd DS (1981) Ten-year experience in renal transplantation for Fabry’s disease. Transplant Proc 13:57–59
Ojo A, Meier-Kriesche HU, Friedman G, Hanson J, Cibrik D, Leichtman A, Kaplan B (2000) Excellent outcome of renal transplantation in patients with Fabry’s disease. Transplantation 69:2337–2339
Donati D, Novario R, Gastaldi L (1987) Natural history and treatment of uremia secondary to Fabry’s disease: an European experience. Nephron 46:353–359
Gantenbein H, Bruder E, Burger HR, Briner J, Binswanger U (1995) Recurrence of Fabry’s disease in a renal allograft 14 years after transplantation. Nephrol Dial Transplant 10:287–289
Peces R (1996) Is there true recurrence of Fabry’s disease in the transplanted kidney (letter)? Nephrol Dial Transplant 11:561
Brady RO, Tallman JF, Johnson WG, Gal AE, Leahy WR, Quirk JM, Dekaban AS (1973) Replacement therapy for inherited enzyme deficiency. Use of purified ceramidetrihexosidase in Fabry’s disease. N Engl J Med 289:9–14
Desnick RJ, Dean KJ, Grabowski G, Bishop DF, Sweeley CC (1979) Enzyme therapy in Fabry disease: differential in vivo plasma clearance and metabolic effectiveness of plasma and splenic alpha-galactosidase A isozymes. Proc Natl Acad Sci U S A 76:5326–5330
Ioannou YA, Zeidner KM, Gordon RE, Desnick RJ (2001) Fabry disease: preclinical studies demonstrate the effectiveness of alpha-galactosidase A replacement in enzyme-deficient mice. Am J Hum Genet 68:14–25
Schiffmann R, Murray GJ, Treco D, Daniel P, Sellos-Moura M, Myers M, Quirk JM, Zirzow GC, Borowski M, Loveday K, Anderson T, Gillespie F, Oliver KL, Jeffries NO, Doo E, Liang TJ, Kreps C, Gunter K, Frei K, Crutchfield K, Selden RF, Brady RO (2000) Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci U S A 97:365–370
Eng CM, Banikazemi M, Gordon RE, Goldman M, Phelps R, Kim L, Gass A, Winston J, Dikman S, Fallon JT, Brodie S, Stacy CB, Mehta D, Parsons R, Norton K, O’Callaghan M, Desnick RJ (2001) A phase 1/2 clinical trial of enzyme replacement in fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet 68:711–722
Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ (2001) Safety and efficacy of recombinant human alpha-galactosidase A—replacement therapy in Fabry’s disease. N Engl J Med 345:9–16
Schiffmann R, Kopp JB, Austin HA 3rd, Sabnis S, Moore DF, Weibel T, Balow JE, Brady RO (2001) Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 285:2743–2749
Moore DF, Altarescu G, Ling GS, Jeffries N, Frei KP, Weibel T, Charria-Ortiz G, Ferri R, Arai AE, Brady RO, Schiffmann R (2002) Elevated cerebral blood flow velocities in Fabry disease with reversal after enzyme replacement. Stroke 33:525–531
Tondel C, Laegreid LM, Hirth A, Houge G, Mansson JE, Sovik O (2004) Intravenous enzyme substitution therapy in children with Fabry’s disease. Tidsskr Nor Laegeforen 123:3388–3390
Illsinger S, Luecke T, Langen H, Das AM (2003) Enzyme replacement therapy in an adolescent with Fabry disease. Eur J Pediatr 162:522–523
Baehner F, Kampmann C, Whybra C, Miebach E, Wiethoff CM, Beck M (2003) Enzyme replacement therapy in heterozygous females with Fabry disease: results of a phase IIIB study. J Inherit Metab Dis 26:617–627
Weidemann F, Breunig F, Beer M, Sandstede J, Turschner O, Voelker W, Ertl G, Knoll A, Wanner C, Strotmann JM (2003) Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease: a prospective strain rate imaging study. Circulation 108:1299–1301
Hajioff D, Enever Y, Quiney R, Zuckerman J, Mackermot K, Mehta A (2003) Hearing loss in Fabry disease: the effect of agalsidase alfa replacement therapy. J Inherit Metab Dis 26:787–794
De Schoenmakere G, Chauveau D, Grunfeld JP (2003) Enzyme replacement therapy in Anderson-Fabry’s disease: beneficial clinical effect on vital organ function. Nephrol Dial Transplant 18:33–35
Breunig F, Weidemann F, Beer M, Eggert A, Krane V, Spindler M, Sandstede J, Strotmann J, Wanner C (2003) Fabry disease: diagnosis and treatment. Kidney Int [Suppl]:S181–185
Acknowledgements
The authors wish to thank Drs. Roscoe Brady and Robert Kleta for critical reviews of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cho, M.E., Kopp, J.B. Fabry disease in the era of enzyme replacement therapy: a renal perspective. Pediatr Nephrol 19, 583–593 (2004). https://doi.org/10.1007/s00467-004-1466-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-004-1466-4