Mutant IDH1 Differently Affects Redox State and Metabolism in Glial Cells of Normal and Tumor Origin

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Stable Transduction of IDH1R132H in Glioblastoma Cells and Immortalized Astrocytes
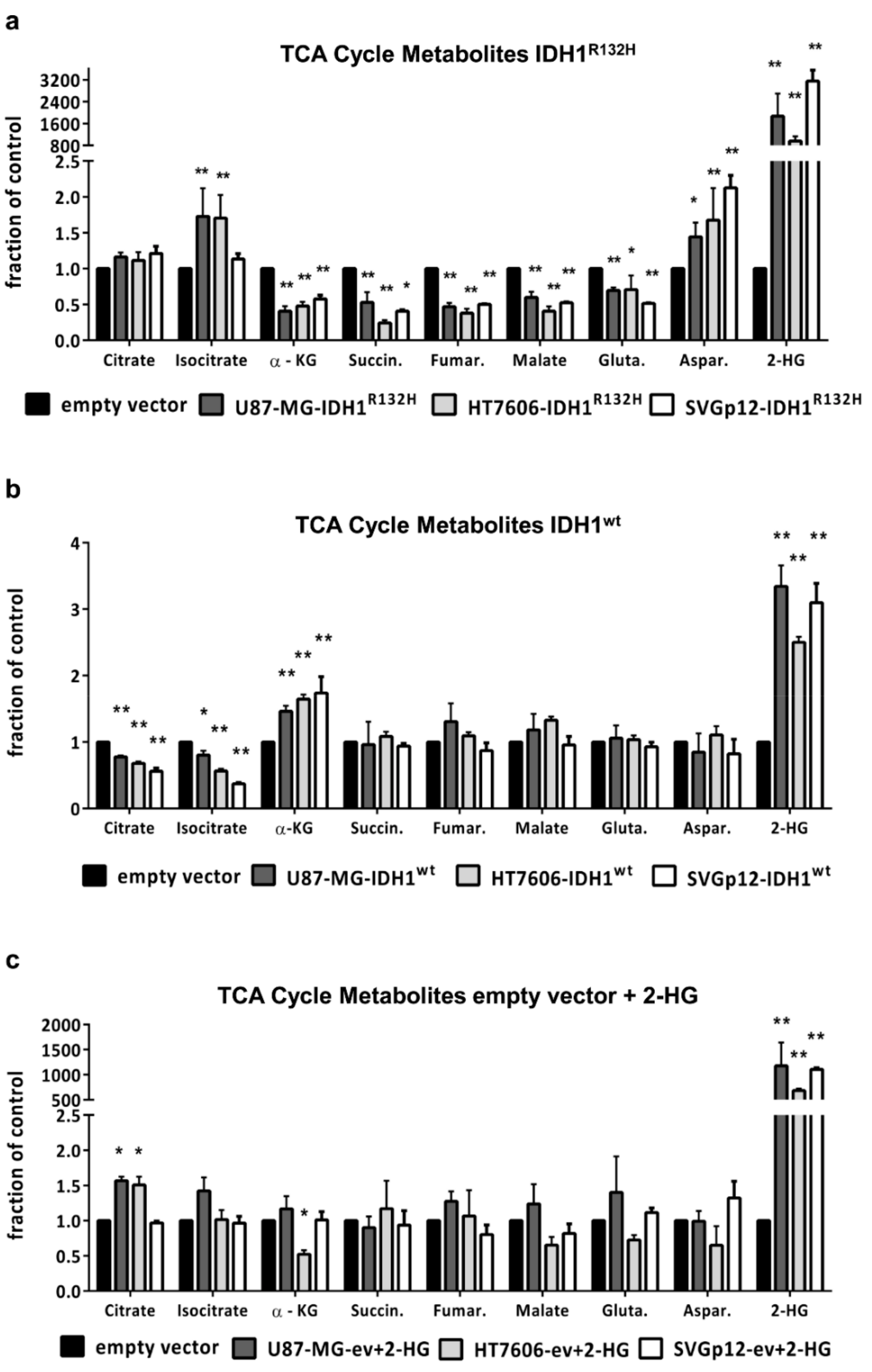
2.2. IDH1R132H but not 2-HG Treatment Alone Leads to Changes in TCA Cycle Metabolites
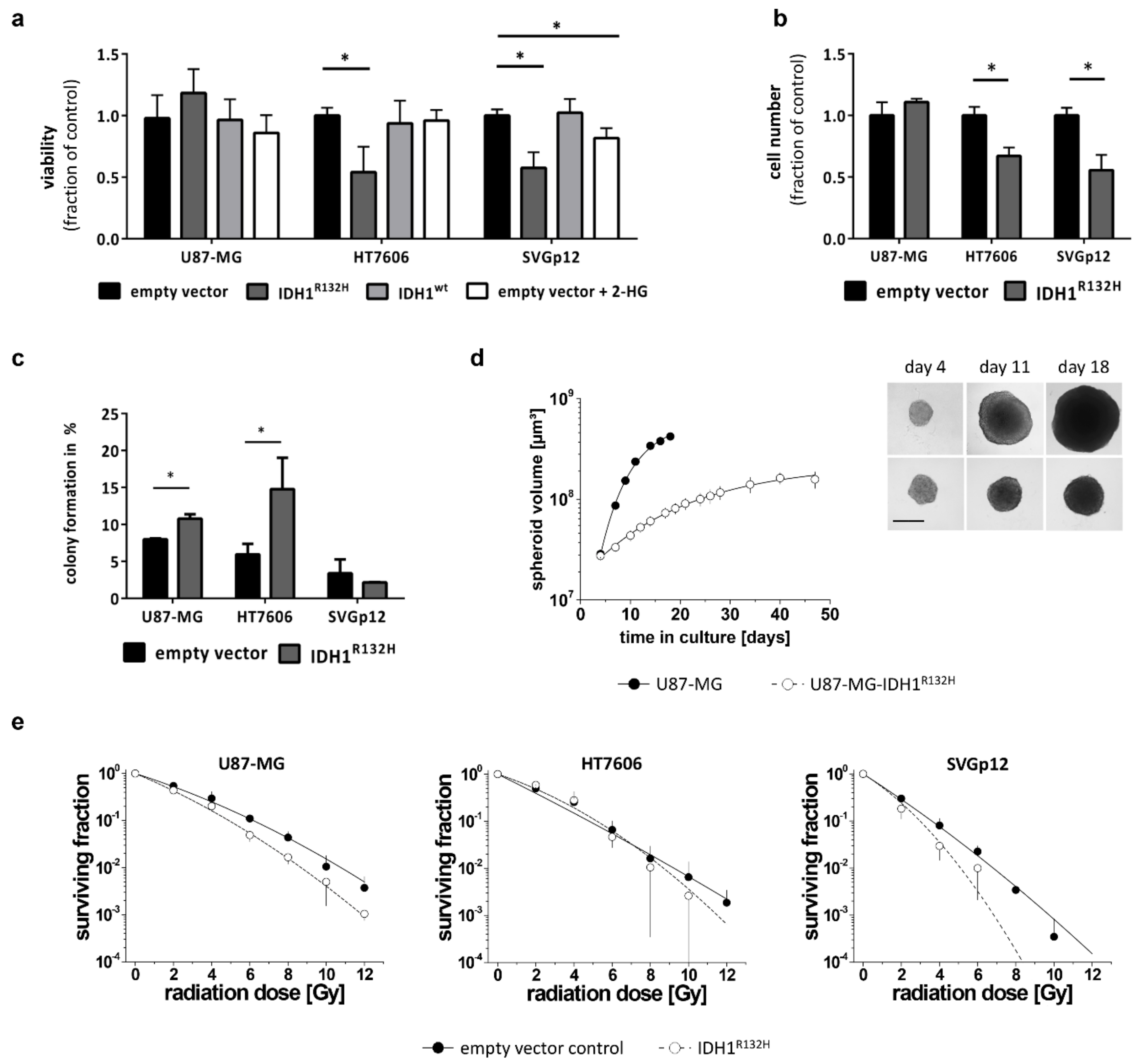
2.3. IDH1R132H Inhibits Growth and Enhances Radio-Sensitivity In Vitro
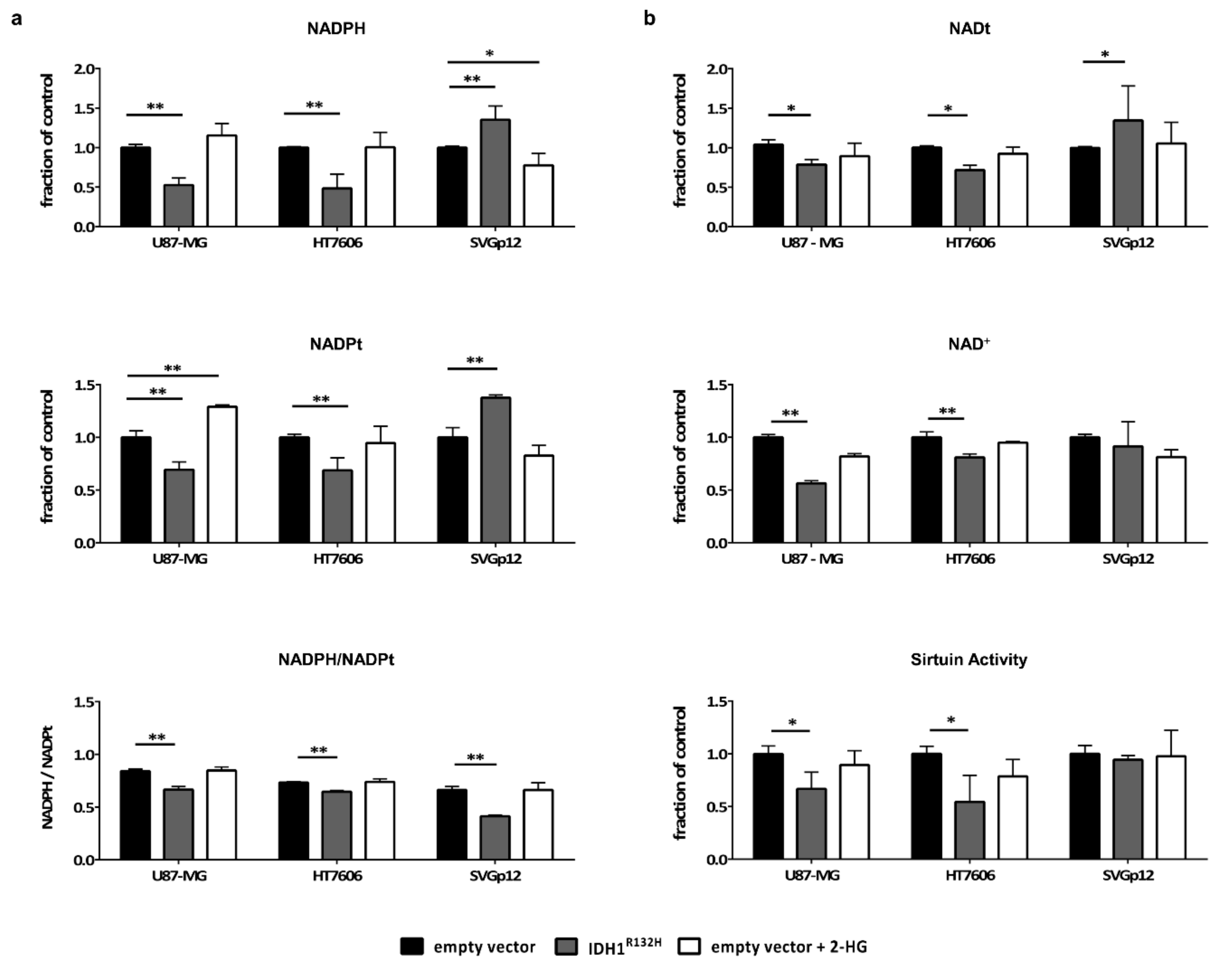
2.4. Intracellular NADPH Levels Significantly Drop in Glioma Cells but not in Astrocytes Upon Transduction with IDH1R132H
2.5. IDH1R132H Leads to a Decrease in NAD+ and the Activity of NAD-Dependent Enzymes in Glioma Cells but not in Astrocytes
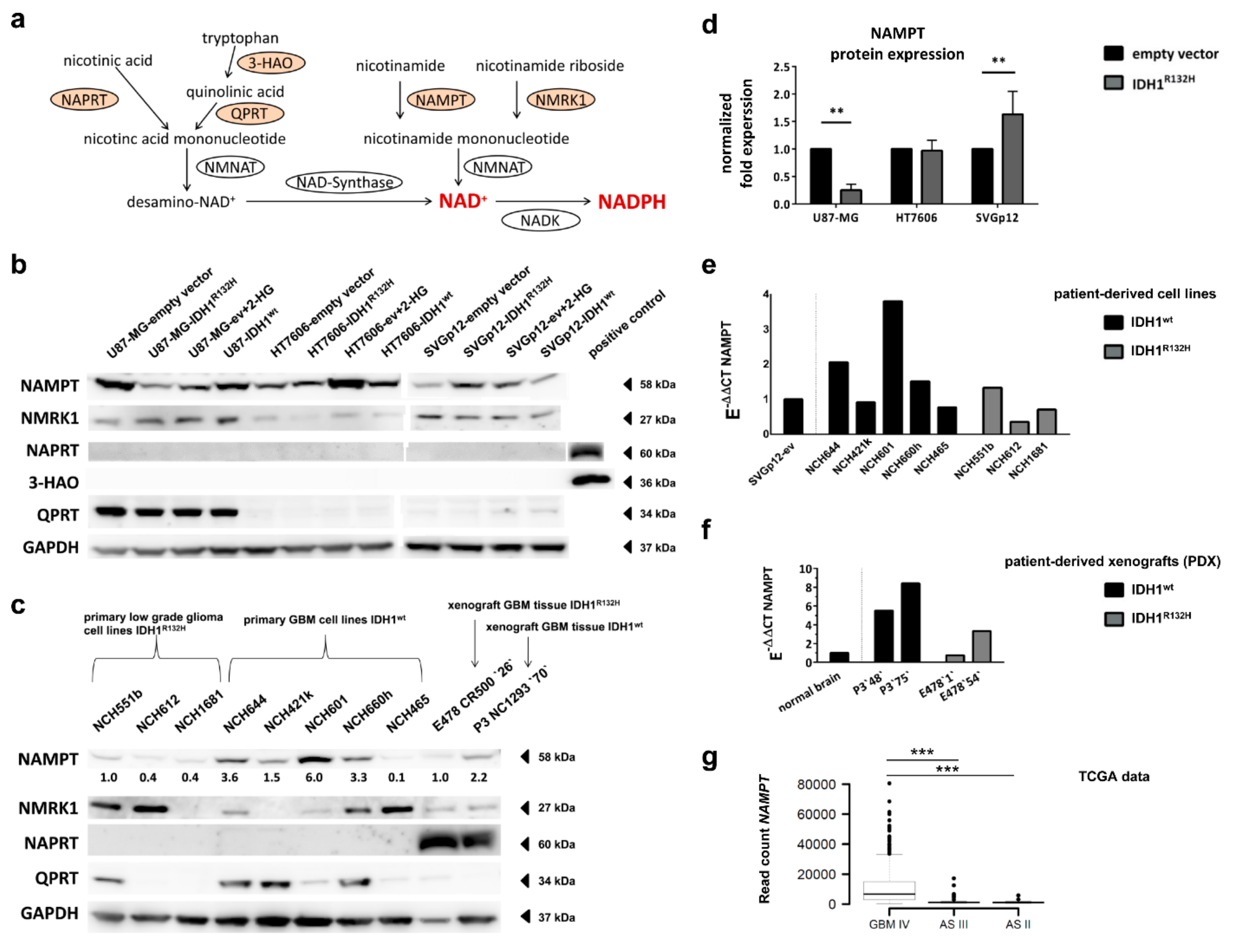
2.6. Expression of NAD+ Synthesis Enzymes Varies between Individual Cell Lines
2.7. Different Effect of IDH1R132H on NAMPT-Expression between Glioma Cells and Astrocytes
2.8. Expression of NAD+ Synthesis Enzymes Varies in Patient-Derived IDH1R132H and IDH1wt Glioma Cells
2.9. IDH1R132H Gliomas Show Lower Expression of NAMPT In Vivo
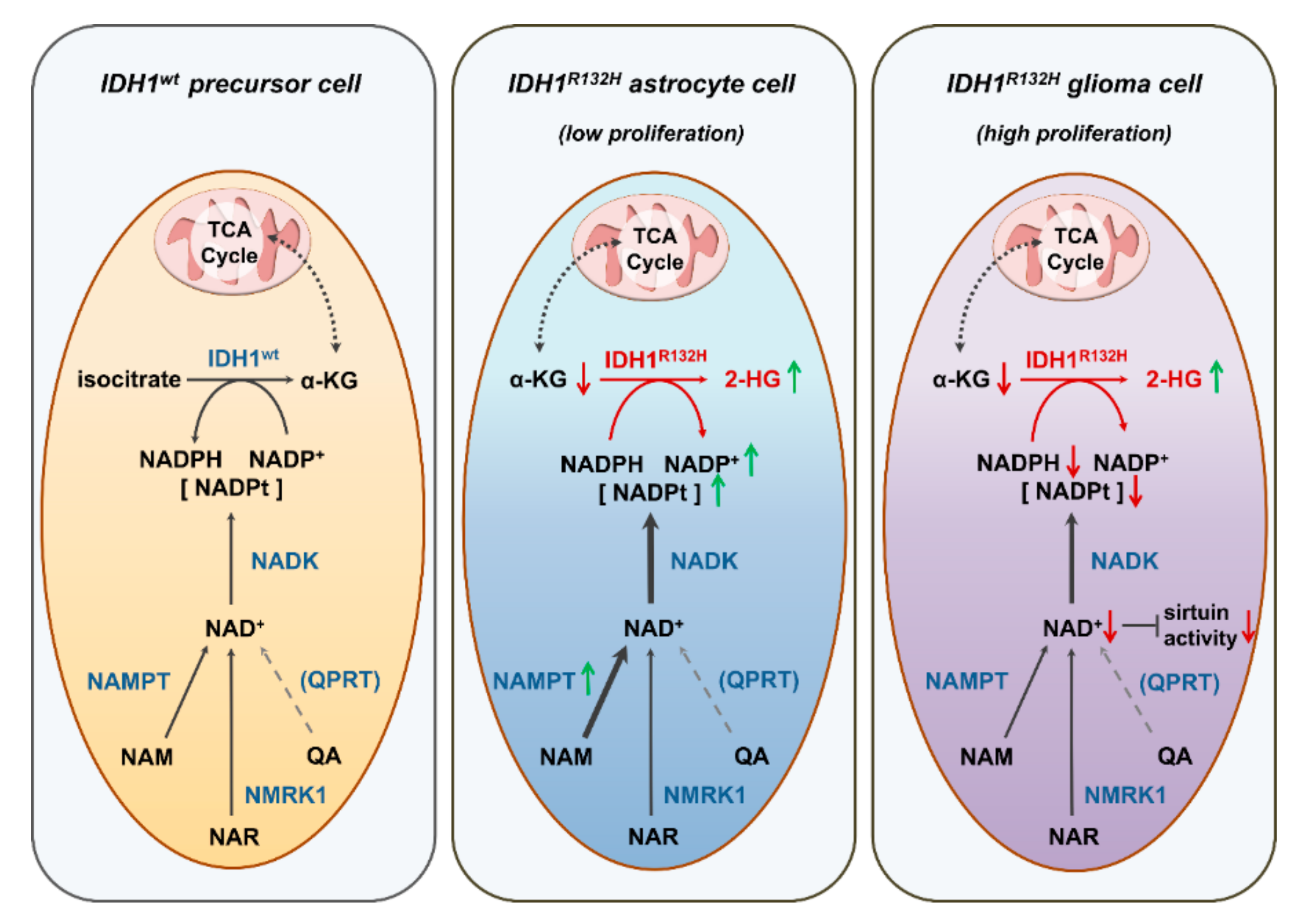
3. Discussion
4. Materials and Methods
4.1. Cells and Cell Culture
4.2. Patient-Derived In Vitro and In Vivo Models
4.3. Plasmid Constructs and Lentivirus Production
4.4. DNA and RNA Extraction
4.5. PCR and Sequencing
4.6. Quantitative Real-Time PCR
4.7. Protein Extraction and Western Blot
4.8. Quantification of TCA Cycle Metabolite Levels Using Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
4.9. Colorimetric Measurement of Cellular NAD and NADPH Levels
4.10. Fluorimetric Measurement of Sirtuin Activity
4.11. Two-Dimensional Growth Assays
4.12. Three-Dimensional Growth Assay
4.13. Clonogenic Survival
4.14. Statistical Analysis
4.15. TCGA Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.S.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [PubMed] [Green Version]
- Beiko, J.; Suki, D.; Hess, K.R.; Fox, B.D.; Cheung, V.; Cabral, M.; Shonka, N.; Gilbert, M.R.; Sawaya, R.; Prabhu, S.S.; et al. IDH1 mutant malignant astrocytomas are more amenable to surgical resection and have a survival benefit associated with maximal surgical resection. Neuro Oncol. 2014, 16, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairncross, J.G.; Wang, M.; Jenkins, R.B.; Shaw, E.G.; Giannini, C.; Brachman, D.G.; Buckner, J.C.; Fink, K.L.; Souhami, L.; Laperriere, N.J.; et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J. Clin. Oncol. 2014, 32, 783–790. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 2016, 18, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Losman, J.-A.; Looper, R.E.; Koivunen, P.; Lee, S.; Schneider, R.K.; McMahon, C.; Cowley, G.S.; Root, D.E.; Ebert, B.L.; Kaelin, W.G. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Kloosterhof, N.K.; Bralten, L.B.C.; Dubbink, H.J.; French, P.J.; van den Bent, M.J. Isocitrate dehydrogenase-1 mutations: A fundamentally new understanding of diffuse glioma? Lancet Oncol. 2011, 12, 83–91. [Google Scholar] [CrossRef]
- Kranendijk, M.; Struys, E.A.; Salomons, G.S.; Van Der Knaap, M.S.; Jakobs, C. Progress in understanding 2-hydroxyglutaric acidurias. J. Inherit. Metab. Dis. 2012, 35, 571–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esmaeili, M.; Hamans, B.C.; Navis, A.C.; Van Horssen, R.; Bathen, T.F.; Gribbestad, I.S.; Leenders, W.P.; Heerschap, A. IDH1 R132H mutation generates a distinct phospholipid metabolite profile in glioma. Cancer Res. 2014, 74, 4898–4907. [Google Scholar] [CrossRef] [Green Version]
- Van Lith, S.A.M.; Navis, A.C.; Verrijp, K.; Niclou, S.P.; Bjerkvig, R.; Wesseling, P.; Tops, B.; Molenaar, R.; van Noorden, C.J.F.; Leenders, W.P.J. Glutamate as chemotactic fuel for diffuse glioma cells: Are they glutamate suckers? Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 66–74. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Duncan, C.G.; Poteet, E.; Winters, A.; Yan, L.J.; Gooden, D.M.; Spasojevic, I.; Boros, L.G.; Yang, S.H.; Yan, H. Cancer-Associated isocitrate dehydrogenase 1 (IDH1) R132H mutation and D-2-hydroxyglutarate stimulate glutamine metabolism under hypoxia. J. Biol. Chem. 2014, 289, 23318–23328. [Google Scholar] [CrossRef] [Green Version]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.V.; Bernard, A.; Hau, A.-C.; et al. Altered metabolic landscape in IDH-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef]
- Bleeker, F.E.; Atai, N.A.; Lamba, S.; Jonker, A.; Rijkeboer, D.; Bosch, K.S.; Tigchelaar, W.; Troost, D.; Van Dertop, W.P.; Bardelli, A.; et al. The prognostic IDH1R132 mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010, 119, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Hirrlinger, J.; Dringen, R. The cytosolic redox state of astrocytes: Maintenance, regulation and functional implications for metabolite trafficking. Brain Res. Rev. 2010, 63, 177–188. [Google Scholar] [CrossRef]
- Richter, S.; Peitzsch, M.; Rapizzi, E.; Lenders, J.W.; Qin, N.; De Cubas, A.A.; Schiavi, F.; Rao, J.U.; Beuschlein, F.; Quinkler, M.; et al. Krebs cycle metabolite profiling for identification and stratification of pheochromocytomas/paragangliomas due to succinate dehydrogenase deficiency. J. Clin. Endocrinol. Metab. 2014, 99, 3903–3911. [Google Scholar] [CrossRef] [Green Version]
- Juratli, T.A.; Peitzsch, M.; Geiger, K.; Schackert, G.; Eisenhofer, G.; Krex, D. Accumulation of 2-hydroxyglutarate is not a biomarker for malignant progression in IDH-mutated low-grade gliomas. Neuro Oncol. 2013, 15, 682–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinrichs, C.N.; Ingargiola, M.; Kaubler, T.; Lock, S.; Temme, A.; Kohn-Luque, A.; Deutsch, A.; Vovk, O.; Stasyk, O.; Kunz-Schughart, L.A. Arginine Deprivation Therapy: Putative Strategy to Eradicate Glioblastoma Cells by Radiosensitization. Mol. Cancer Ther. 2018, 17, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, N.; Niere, M.; Ziegler, M. NAD kinase levels control the NADPH concentration in human cells. J. Biol. Chem. 2007, 282, 33562–33571. [Google Scholar] [CrossRef] [Green Version]
- Gray, J.P.; Alavian, K.N.; Jonas, E.A.; Heart, E.A. NAD kinase regulates the size of the NADPH pool and insulin secretion in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 2012, 303, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Chiarugi, A.; Dolle, C.; Felici, R.; Ziegler, M.; Dölle, C.; Felici, R.; Ziegler, M.; Dolle, C.; Felici, R.; Ziegler, M. The NAD metabolome--a key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef]
- Lee, D.H. Sirt1 as a New Therapeutic Target in Metabolic and Age-Related Diseases. Chonnam Med. J. 2010, 46, 67–73. [Google Scholar] [CrossRef]
- Ohka, F.; Ito, M.; Ranjit, M.; Senga, T.; Motomura, A.; Motomura, K.; Saito, K.; Kato, K.; Kato, Y.; Wakabayashi, T.; et al. Quantitative metabolome analysis profiles activation of glutaminolysis in glioma with IDH1 mutation. Tumor. Biol. 2014, 35, 5911–5920. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef] [Green Version]
- Khurshed, M.; Molenaar, R.J.; Lenting, K.; Leenders, W.P.; van Noorden, C.J.F. In silico gene expression analysis reveals glycolysis and acetate anaplerosis in IDH1 wild-type glioma and lactate and glutamate anaplerosis in IDH1-mutated glioma. Oncotarget 2017, 8, 49165. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.J.; Shih, H.A.; Andronesi, O.C.; Cahill, D.P. Isocitrate dehydrogenase-mutant glioma: Evolving clinical and therapeutic implications. Cancer 2017, 123, 4535–4546. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, H.; Tanaka, K.; Sasayama, T.; Irino, Y.; Sato, N.; Takeuchi, Y.; Kyotani, K.; Mukasa, A.; Mizukawa, K.; Sakata, J.; et al. Diagnostic value of glutamate with 2-hydroxyglutarate in magnetic resonance spectroscopy for IDH1 mutant glioma. Neuro Oncol. 2016, 18, 1559–1568. [Google Scholar] [PubMed] [Green Version]
- Bralten, L.B.C.; Kloosterhof, N.K.; Balvers, R.; Sacchetti, A.; Lapre, L.; Lamfers, M.; Leenstra, S.; De Jonge, H.; Kros, J.M.; Jansen, E.E.W.; et al. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Ann. Neurol. 2011, 69, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Radivoyevitch, T.; Maciejewski, J.P.; van Noorden, C.J.F.; Bleeker, F.E. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.; Güttler, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. IDH1R132H mutation causes a less aggressive phenotype and radiosensitizes human malignant glioma cells independent of the oxygenation status. Radiother. Oncol. 2015, 116, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Botman, D.; Smits, M.A.; Hira, V.V.; Van Lith, S.A.; Stap, J.; Henneman, P.; Khurshed, M.; Lenting, K.; Mul, A.N.; et al. Radioprotection of IDH1-mutated cancer cells by the IDH1-mutant inhibitor AGI-5198. Cancer Res. 2015, 75, 4790–4802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, D.R.; Dresser, J.; Wilder-Romans, K.; Parsels, J.D.; Zhao, S.G.; Davis, M.; Zhao, L.; Kachman, M.; Wernisch, S.; Burant, C.F.; et al. Glioblastoma Therapy Can Be Augmented by Targeting IDH1-Mediated NADPH Biosynthesis. Cancer Res. 2017, 77, 960–970. [Google Scholar] [CrossRef] [Green Version]
- Kessler, J.; Hohmann, T.; Güttler, A.; Petrenko, M.; Ostheimer, C.; Hohmann, U.; Bache, M.; Dehghani, F.; Vordermark, D. Radiosensitization and a Less Aggressive Phenotype of Human Malignant Glioma Cells Expressing Isocitrate Dehydrogenase 1 (IDH1) Mutant Protein: Dissecting the Mechanisms. Cancers 2019, 11, 889. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, K.; Kawai, S.; Koshimizu, M.; Murata, K. NADPH regulates human NAD kinase, a NADP +-biosynthetic enzyme. Mol. Cell. Biochem. 2011, 355, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.; Roulston, A.; Bélec, L.; Billot, X.; Marcellus, R.; Bédard, D.; Bernier, C.; Branchaud, S.; Chan, H.; Dairi, K.; et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: Strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell. Biol. 2009, 29, 5872–5888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Oezen, I.; Opitz, C.A.; Radlwimmer, B.; Von Deimling, A.; Ahrendt, T.; Adams, S.; Bode, H.B.; Guillemin, G.J.; Wick, W.; et al. The endogenous tryptophan metabolite and NAD+ precursor quinolinic acid confers resistance of gliomas to oxidative stress. Cancer Res. 2013, 73, 3225–3234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bürkle, A. Poly(ADP-ribose): The most elaborate metabolite of NAD+. FEBS J. 2005, 272, 4576–4589. [Google Scholar] [CrossRef]
- Inoue, T.; Hiratsuka, M.; Osaki, M.; Oshimura, M. The molecular biology of mammalian SIRT proteins: SIRT2 in cell cycle regulation. Cell Cycle 2007, 6, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.I.; Guarente, L. Ten years of NAD-dependent SIR2 family deacetylases: Implications for metabolic diseases. Trends Pharmacol. Sci. 2010, 31, 212–220. [Google Scholar] [CrossRef]
- Dang, W. The controversial world of sirtuins. Drug Discov. Today Technol. 2014, 12, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Hendruschk, S.; Wiedemuth, R.; Aigner, A.; Töpfer, K.; Cartellieri, M.; Martin, D.; Kirsch, M.; Ikonomidou, C.; Schackert, G.; Temme, A. RNA interference targeting survivin exerts antitumoral effects in vitro and in established glioma xenografts in vivo. Neuro Oncol. 2011, 13, 1074–1089. [Google Scholar] [CrossRef] [Green Version]
- Pusch, S.; Krausert, S.; Fischer, V.; Balss, J.; Ott, M.; Schrimpf, D.; Capper, D.; Sahm, F.; Eisel, J.; Beck, A.-C.; et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017, 133, 629–644. [Google Scholar] [CrossRef]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef]
- Campos, B.; Gal, Z.; Baader, A.; Schneider, T.; Sliwinski, C.; Gassel, K.; Bageritz, J.; Grabe, N.; von Deimling, A.; Beckhove, P.; et al. Aberrant self-renewal and quiescence contribute to the aggressiveness of glioblastoma. J. Pathol. 2014, 234, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Navis, A.C.; Niclou, S.P.; Fack, F.; Stieber, D.; van Lith, S.; Verrijp, K.; Wright, A.; Stauber, J.; Tops, B.; Otte-Holler, I.; et al. Increased mitochondrial activity in a novel IDH1-R132H mutant human oligodendroglioma xenograft model: In situ detection of 2-HG and alpha-KG. Acta Neuropathol. Commun. 2013, 1, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, N.; Michen, S.; Tietze, S.; Topfer, K.; Schulte, A.; Lamszus, K.; Schmitz, M.; Schackert, G.; Pastan, I.; Temme, A. Engineering NK Cells Modified With an EGFRvIII-specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1alpha-secreting Glioblastoma. J. Immunother. 2015, 38, 197–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Spheroid-based drug screen: Considerations and practical approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef]
- Ingargiola, M.; Runge, R.; Heldt, J.-M.; Freudenberg, R.; Steinbach, J.; Cordes, N.; Baumann, M.; Kotzerke, J.; Brockhoff, G.; Kunz-Schughart, L.A. Potential of a Cetuximab-based radioimmunotherapy combined with external irradiation manifests in a 3-D cell assay. Int. J. Cancer 2014, 135, 968–980. [Google Scholar] [CrossRef] [Green Version]
- Dittfeld, C.; Dietrich, A.; Peickert, S.; Hering, S.; Baumann, M.; Grade, M.; Ried, T.; Kunz-Schughart, L.A. CD133 expression is not selective for tumor-initiating or radioresistant cell populations in the CRC cell line HCT-116. Radiother. Oncol. 2010, 94, 375–383. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biedermann, J.; Preussler, M.; Conde, M.; Peitzsch, M.; Richter, S.; Wiedemuth, R.; Abou-El-Ardat, K.; Krüger, A.; Meinhardt, M.; Schackert, G.; et al. Mutant IDH1 Differently Affects Redox State and Metabolism in Glial Cells of Normal and Tumor Origin. Cancers 2019, 11, 2028. https://doi.org/10.3390/cancers11122028
Biedermann J, Preussler M, Conde M, Peitzsch M, Richter S, Wiedemuth R, Abou-El-Ardat K, Krüger A, Meinhardt M, Schackert G, et al. Mutant IDH1 Differently Affects Redox State and Metabolism in Glial Cells of Normal and Tumor Origin. Cancers. 2019; 11(12):2028. https://doi.org/10.3390/cancers11122028
Chicago/Turabian StyleBiedermann, Julia, Matthias Preussler, Marina Conde, Mirko Peitzsch, Susan Richter, Ralf Wiedemuth, Khalil Abou-El-Ardat, Alexander Krüger, Matthias Meinhardt, Gabriele Schackert, and et al. 2019. "Mutant IDH1 Differently Affects Redox State and Metabolism in Glial Cells of Normal and Tumor Origin" Cancers 11, no. 12: 2028. https://doi.org/10.3390/cancers11122028