
Abstract
The human immunodeficiency virus (HIV) had spread unrecognized in the human population as sexually transmitted disease and was finally identified by its disease AIDS in 1981. Even after the isolation of the causative agent in 1983, the burden and death rate of AIDS accelerated worldwide especially in young people despite the confection of new drugs capable to inhibit virus replication since 1997. However, at least in industrialised countries, this trend could be reversed by the introduction of combination therapy strategies. The design of new drugs is on going; besides the inhibition of the three enzymes of HIV for replication and maturation (reverse transcriptase, integrase and protease), further drugs inhibits fusion of viral and cellular membranes and virus maturation. On the other hand, viral diagnostics had been considerably improved since the emergence of HIV. There was a need to identify infected people correctly, to follow up the course of immune reconstitution of patients by measuring viral load and CD4 cells, and to analyse drug escape mutations leading to drug resistance. Both the development of drugs and the refined diagnostics have been transferred to the treatment of patients infected with hepatitis B virus (HBV) and hepatitis C virus (HCV). This progress is not completed; there are beneficial aspects in the response of the scientific community to the HIV burden for the management of other viral diseases. These aspects are described in this contribution. Further aspects as handling a stigmatising disease, education of self-responsiveness within sexual relationships, and ways for confection of a protective vaccine are not covered.
Similar content being viewed by others


Introduction
The human immunodeficiency virus (HIV) was isolated in 1983 for the first time just 2 years after the description of a new disease affecting especially young homosexually active men [1]; originally it has been named lymphadenopathy associated virus (LAV) [2] and human T lymphotropic virus type III (HTLV-III) [3], or got other names as AIDS related virus (ARV) [4]. HIV is a member of the lentiviruses within the retrovirus family. Lentiviruses are prevalent in plenty mammalian species as horse (equine infectious anaemia virus), sheep (visna maedi virus), cattle (bovine immunodeficiency virus), cat (feline immunodeficiency virus), and finally in nearly all monkey species (simian immunodeficiency virus, SIV). Naturally, monkeys living in Africa, Asia and South America are infected with the simian T leukaemia virus (STLV) while lentivirus infected monkeys are only found in Africa [5], indicating that in terms of viral evolution, HIV is younger than the related retrovirus STLV/HTLV. Monkeys infected with SIV, which is endogenous in that species, will not develop immunodeficiency; in contrast, SIV transmitted within monkey species can lead to an acute illness associated with immunodeficiency and AIDS like symptoms [6, 7].
HIV is divided into two types, HIV-1 and HIV-2. HIV-1 consists of the groups M, N and O, HIV-2 of the groups A to G, which were formerly named subtypes. On the molecular basis, HIV-1 group M is nearly identical to SIVcpz, virus clades that are found in chimpanzees (Pan troglodytes) in Central Africa, and HIV-1 group O viruses have been detected in the faeces of three wild living gorillas in Cameroon [8], while HIV-2 is identical to SIV from sooty mangabey monkeys (Cercopithecus atys—SIV smm) [5]. It seems that in monkeys, host and virus have adapted over time to equilibrium in such a way that the virus replicates to high amounts, but no longer kills the host. In humans both HIV-1 and HIV-2 cause immunodeficiency (AIDS); this usually occurs in HIV-1 and HIV-2 infected but untreated patients after 10 and 15 years, respectively. The lacking pathogenicity in monkeys compared to the high pathogenicity in humans and historical analyses showed an increase of the anti-HIV seroprevalence in stored plasma samples in Central-Africa between 1950 and 1970 [4, 9, 10] and the lacking or low prevalence in pygmies in Cameroon [11] are further arguments that HIV was introduced to humans only ‘recently’, most probably around the time of the beginning of World War 2 or perhaps slightly earlier [12].
The heterogeneity of HIV is easily shown in group M, which is most prevalent in the world and is subdivided into the subtypes A–K; subtypes E and I are missing, because these were finally identified as recombinant forms. Actually, frequently circulating recombinant form viruses (CRF) are classified in CRF01–CRF43 and U for unknown subtype [13, 14]; the actual list is available at http://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html. A recombination event occurs frequently when one cell is infected with two HIV strains; it is assumed that one of 400 HIV secreted from a double infected cell is a recombinant virus. As far as analysed, SIVcpz from chimpanzees is by itself a stable recombinant form, in which the gag and the pol gene region originate from the red capped mangabey monkeys (Cercopithecus torquatus) SIV, and the env gene region from the white nose or greater spot nose monkeys (Cercopithecus nictitans) SIV [15–17]. The origin of the long terminal repeat region (LTR) of the chimpanzee virus is still unclear. From an evolutionary point of view, the diversification of the chimpanzee species in West Central Africa was driven by separation through the big rivers [16], leading to the development of the species Pan troglodytes troglodytes, Pan t schweinfurthii in the Eastern Central part, and Pan t vellerosus in the North-West Central part [17, 18], which might have happened around 100,000–200,000 years ago. All these species actually carry a similar SIV, thus a recombinant virus might have led to a stable infection in chimpanzees in ancient times before further diversification. All SIVcpz seem to be transmissible to humans.
The dynamic of HIV transmission is driven by two forces leading to a worldwide spread: sexual intercourse and blood contamination. The latter occurs now mostly by intravenous drug consumption in the western world, and still by blood transfusion in combination with a highly increasing HIV incidence in developing countries. Before the beneficial period of antiretroviral treatment, most of the HIV infected patients were dying with typical symptoms of AIDS caused by opportunistic infections, neoplasia, marasmus associated with diarrhoea and/or encephalopathy. This death rate in young people was a tremendous stimulus to search for substances to improve antiretroviral therapy (ART), which started in 1989 using zidovudine monotherapy, and became sufficiently effective after the introduction of protease inhibitors in 1996 (Table 1) resulting in the principle of highly active antiretroviral therapy (HAART) [19].
The diagnostic task resulting from the spread of HIV was to identify infected people as precisely as possible by reliable antibody tests and to monitor the status of patients by laboratory methods. Laboratory markers like viral load and CD4 cell count are useful to get information on the fitness of the virus and the residual power of the immune system, respectively. Drug resistance determination was a prompt step following ART, taking into account that the mutation rate of HIV is relatively high with approximately 1 in 10,000, similar to that of plenty other RNA viruses [4, 10].
Improvement of diagnostics for HIV antibody, antigen and nucleic acid detection
HIV-antibody testing
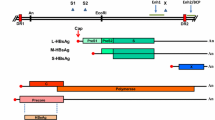
In 1985, the first HIV antibody ELISAs were available and it was evident from the beginning that numerous test results had been false positive, especially when testing pregnant women. Confirmatory tests were needed, which were based on immunofluorescence, since HIV could be grown nicely on T-cell lines [20, 21], and western blot strips, which used more or less highly purified antigenic material derived from virus concentrated from supernatant of infected cells. A consecutive procedure was the production of line immunoassays with antigenic material purified after cloning individual HIV proteins (Fig. 1).

Scheme of the algorithm of antibody, antigen and viral nucleic acid testing in the diagnostics of HIV, HCV and HBV infection. When the diagnostic window is closed and the initial antibody testing result is negative virus infection is excluded, therefore further data are not documented in the algorithm. When patients have to be treated dependent predominantly on their clinical status, which is not cited in this scheme, a circle of drug application, drug resistance testing and viral load and CD4 cells determination in HIV infected patients, and viral load and liver enzyme testing, as ALT and AST, in hepatitis patients has to be pursued. Permanent treatment is obligatory in AIDS patients and treatment in hepatitis patients occurs until improvement or final failure is obvious
ELISA refinement went through different steps including peptides as antigen, followed by using material from HIV-1 group M, group O and HIV-2 produced by recombinant techniques, and switching from the indirect ELISA format to the double antigen format, which allowed the simultaneous detection of IgM and IgG. Today’s tests are combination tests, which detect HIV antibody and the p24-antigen simultaneously [4, 22].
Detection of viral components by p24-antigen testing
To get information on the viral burden in AIDS patients, an antigen assay was developed in the sandwich ELISA format using the core protein p24 in 1988; around 1,000 p24 molecules build up the conical core of one HIV particle. As a result of the benefit for the HIV diagnostic, a core-antigen assay was developed for quantitative determination of HCV in plasma 11 years later in 1999 [23, 24].
P24-antigen assays were very quickly replaced when nucleic acid testing allowed the quantitative determination of the HIV-RNA viral load in plasma. In addition, it became evident that p24-antigen and viral load did not correlate, since p24 is shed in huge amounts from the surface of some infected cells [25].
Viral load tested by nucleic acid testing (NAT)
Different methods have been applied for HIV-1 nucleic acid detection such as PCR, NASBA, TMA, b-DNA, and finally Taqman-PCR as a further refinement [26]. The commercially available tests have high sensitivity and high throughput performance, and are not only used to monitor patients, but also to increase safety of the blood supply by the ability to detect HIV-RNA during early seroconversion [27, 28]. To reach a high sensitivity, concentration of HIV by ultracentrifugation was introduced as routine analysis.
Taken together, strategies and experiences made in HIV testing had been transferred to other infectious agents: to hepatitis C virus (HCV) concerning immunoblot as line immunoassay, core antigen detection and nucleic acid testing, as well as to hepatitis B virus (HBV) concerning viral load quantification and drug resistance determination (Fig. 1). Additionally, experience gained from HIV has been transferred to the diagnostics of EBV, CMV, and further viruses, to bacteria as Borrelia, Treponema pallidum, to Toxoplasma and further infectious agents concerning confirmation of ELISA or agglutination test reactivity by immunoblot. Nucleic acid testing has nearly completely replaced virus isolation for all viruses and older methods of virus detection like immunofluorescence microscopy or antigen assays, since NAT is faster, more sensitive and even cheaper.
Drug resistance testing
HIV has a high mutation rate due to the infidelity of the reverse transcriptase. Because of a not fully suppressive ART, HIV mutants are selected which replicate in the presence of the drug; hence, the drug is bound insufficiently to the enzyme. The interaction of the drug and the corresponding enzyme is associated with defined amino acids (key amino acids); both the wild type and the respective mutant could be identified by nucleic acid sequencing of the respective viral gene [29]. Generally, two methods, phenotypic and genotypic testing, are available; for several reasons genotypic testing, which is based on nucleic acid sequencing, is more favourable.
Drug resistance testing has now been introduced to HBV and HCV diagnostics (Fig. 1) and will be extended further to those viral diseases which are treatable with effective antiviral substances, such as neuraminidase inhibitors for influenza virus A and B especially concerning H5N1 [30, 31]. Resistance testing is still of minor value for viruses that are permanently hidden in human cells such as herpes virus.
A further step forward was prediction of drug susceptibility by resistance determination before starting treatment. Analysis of subtype and genotype was based on the experience gathered by HPV [31]. Routine genotype analysis has been extended now to HIV, HBV and HCV, and finally is used for epidemiological studies of rota-, noro-, adeno-, measles-virus, and other viruses [32, 33].
In this sense, methods introduced in routine HIV diagnostics did smoothen the way for other infectious agents resulting in a significant benefit for treating and monitoring patients.
Improvement of antiretroviral therapy and designing new drugs
Drug design for inhibition of virus replication started after successful application of acyclovir and its inhibition of the HSV polymerase (thymidine kinase), which was extend later to beta-herpes virus as cytomegalovirus (CMV) with ganciclovir [34]. Suffering and death of AIDS patients on the one hand and knowledge of the structure of the virus particle, replication cycle and three-dimensional configuration of the enzymes on the other hand were a driving force for the design of drugs able to inhibit viral growth and thus reduce the pathogenic action of HIV [35].
HIV reverse transcriptase (RT)
The enzyme transcribes viral RNA in viral DNA, which is incorporated into the human cell genome by the action of the viral integrase. The first drug applied for RT inhibition was azidothymidine/zidovudine (AZT) (Table 1), a nucleoside analogue that leads to chain termination of the newly synthesized DNA strand after incorporation. The whole class was named nucleoside reverse transcriptase inhibitors (NRTI). Other substances belonging to the NRTIs are didanosine (ddI), zalcitabine (ddC; no longer distributed), stavudine (d4T), lamivudine (3TC), emtricitabine (FTC), and abacavir (ABC) (Table 1). To avoid rapid selection of resistant viruses, the combination of appropriate drugs for treatment is necessary—this lesson was learned within the first year of antiretroviral therapy [19, 35].
Inhibition of the enzyme by non-nucleoside reverse transcriptase inhibitors (NNRTI) was a consequence of the exact mapping of the three-dimensional structure of the reverse transcriptase including the RNase H domain. Presently used drugs are efavirenz (EFV), nevirapine (NVP), etravirine (ETR), and to a smaller extent delavirdine (DLV) (Table 1). Although the NNRTIs are potent drugs in antiretroviral combination treatment, they have a relatively low genetic barrier, which means a rapid selection of drug resistance associated mutations under non-suppressive ART; except for ETR only one amino acid mutation is sufficient to cause complete resistance.
A further step to RT inhibition was the introduction of tenofovir disoproxil fumarate (TDF) a nucleotide reverse transcriptase inhibitor (NtRTI) in 2001. In contrast to the above-mentioned NRTIs, TDF is already monophosphorylated, thus it is activated avoiding the first phosphorylation step [35].
One major message learned from treating HIV infected patients was that besides the knowledge of the action of the drugs, patient’s compliance, adherence and fidelity to the drug administration scheme was highly important for effectiveness of treatment; for example, a once daily is favourable to a twice daily administration.
HBV polymerase
Knowledge of drug interference with the HIV-RT was transferred to HBV. Second DNA strand synthesis completion of HBV can be inhibited by substances known from HIV therapy like 3TC, FTC, and TDF as well as by drugs approved only for HBV treatment like adefovir, entecavir, and telbivudine (Table 1) [36]. All these drugs act within the active enzyme pocket of the HBV polymerase, which contains among other amino acids the motif YMDDV (YMDDL in HIV). Treatment of chronic HBV infection has been considerably improved by the HIV antiretrovirals. Since drug dosage is lower in HBV treatment compared to HIV treatment, side effects are smaller, and thus therapy is more easily accepted by the patient. Studies on combination therapy of the cited drugs are under way, and a further improvement is expected [37].
As observed in the genome of HIV, mutations within the genome of HBV resulting in drug resistance were selected during insufficient treatment, too. For example, mutations at positions rtL180 and rtM204, the latter located in the YMDDV motif, were selected after 3TC treatment [36]. However, mutations in HBV occur later than in HIV since the genome is circular with overlapping reading frames, and therefore mutations in the HBV DNA–RNA polymerase have to be tolerated in the HBV core protein as well.
Inhibition of the HCV replicase
The enzyme responsible for HCV replication is an RNA dependent RNA polymerase encoded from the NS5B gene region. Substances named nucleoside based RNA replicase inhibitors (NRRI) have been designed, for example, valopicitabine, 2-O-methylcytidine, 2-C-methylcytidine, and 2-C-methylguanosine [34]. First clinical trials showed that administration of valopicitabine reduced the HCV viral load by 1.2 log10 after 2 weeks. Additionally, non-nucleoside RNA replicase inhibitors (NNRRI) have been confected; however, benzimidazole-5-carboxyamide and indol-N-acetamide derivatives are currently not applicable due to their toxicity [35, 38].
HIV Protease (PR)
The HIV PR monomer is a molecule consisting of 99 amino acids. The first protease inhibitors (PI) became broadly available in 1997, after the three-dimensional structure of the dimer forming molecule of the PR was elucidated (Table 1). Chemical configurations of substances have been designed, with the background that most of the human proteases are serine-proteases while the HIV PR is an aspartate protease. Drugs like saquinavir (SQV), indinavir (IDV), nelfinavir (NFV), ritonavir (RTV), fos-amprenavir (FPV), lopinavir (LPV), atazanavir (ATZ), tipranavir (TPV), and darunavir (DRV) are currently available (Table 1). Administration of these substances in combination with RT inhibitors led to a sustained and profound decline of the viral load in treated patients [4, 19].
Three additional items have been learned from treatment with protease inhibitors (PRI). The inhibition of the cytochrome oxidase system P450 (CYP 3A4) by PRIs, but especially by RTV, blocks the decomposition of the enzyme and thus prolongs the half-life of some drugs, while it does not interfere with the elimination of others. Based on this observation, most of the PRIs are combined with RTV to elevate the drug level and to enhance the inhibition of the PR; consequently, RTV is currently used as a pharmacokinetic booster only. As one example, NFV acts without interference of the cytochrome oxidase system.
The second point learned was the induction of lipodystrophy. Although aspartate proteases are seldom in the human body, interference of PRIs in genetically predisposed subjects leads to lipodystrophy. The syndrome is associated with fat shrinkage on the extremities, and fat accumulation on the neck, shoulders and the abdomen after several months or even several years of treatment. This kind of side effect is individual, time dependent and sometimes irreversible [39]. Lipodystrophy is as well induced by some NRTI and partially enhanced by the combination of NRTI with PRI.
The third point is that the protease is a small molecule. Some of the amino acid mutations caused by one PRI may be associated with resistance to several other PRI, for example, a point mutation at position 84 is associated with resistance to IDV, RTV, and FPV. This observation led to rapid development of new drugs like ATV, TPV, and DRV (Table 1), which might be still active when some of the key amino acids are mutated.
A further benefit originating from the development of PRI is the observation that RTV, NFV and SQV inhibit the intracellular growth of Leishmania [40, 41].
Inhibition of the HCV protease
The success of a sustained viral load reduction by the introduction of PRIs for treatment of HIV was the basis of designing new protease inhibitors for HCV treatment, for both the NS3/NS4A serine protease and the NS2/3 cysteine protease, which cleave the polyprotein precursor. Active substances are ciluprevir (BLIN 2061), which reduced the viral load by 2–3 log10, telaprevir (VX-950), boceprevir (SCH 503054) and SCH446211. Their clinical use is reserved to ongoing studies. Key amino acid changes causing resistance are N168 for ciluprevir (discontinued in 2005), and V36, T54, R155, A156 and D168 for telaprevir [38, 42]. Telaprevir can be combined with pegylated interferon alpha to suppress HCV production to undetectable levels [43, 44].
HIV integrase inhibitors
The integrase molecule consists of around 250 amino acids, is highly conserved between HIV subtypes, and the enzyme sticks to the HIV nucleic acid forming dimers and tetramers. Besides an integrase activity, the protein exhibits an exonuclease activity. Such an enzyme does not exist in viruses like HBV and HCV and not in human cells. Currently, raltegravir (RAL) and elvitegravir (EVG) (Table 1) have been evaluated in clinical studies [45], and RAL has recently been approved. In contrast to EVG, which has to be taken together with RTV, RAL is eliminated without involvement of the cytochrome oxidase P450 system [46], and until now, no severe side effects have been reported in clinical trials for both substances. As described above for the NNRTIs, one amino acid mutation is linked to a significant loss of antiviral activity; however, the genetic barrier is not as low as for the NNRTIs. Key amino acid mutations for RAL are located at positions Y143, Q148, and N155, but are generally accompanied by secondary mutations such as G140S, and E138A/K. The key amino acids causing resistance to EVG are T66I, E92Q, Q148 K/R, N155H, and to a minor degree G140S, S147G, and Q148H. To preserve the power of these drugs integrase inhibitors can only be applied within an effective combination therapy and they are active against HIV-1 and HIV-2 [35, 45, 47, 48].
Viral entry—fusion inhibitors
To enter the human cell HIV attaches to the CD4 molecule followed by coreceptor binding, and thus initiates the fusion process. The first steps are mediated by the envelope proteins gp120 and gp41. The N-terminal part of gp41 forms a bundle by its two heptad repeat domains (HR1 and HR2), and the gp120 trimer forms a pore through which the hydrophobic N-terminal part of the gp41 trimer molecule inserts into the lipid layer of the cell membrane. Confluence of the viral lipid envelope and the plasma membrane brings the envelope-stripped viral particle to the cytoplasm and terminates the fusion process. When 30-mer peptides, structurally identical to the HR2 region, are attached to gp41, fusion is no longer possible. Based on this knowledge, a 36 amino acid peptide (T-20) was designed, which binds in part to the HR2 domain. When administered to patients, T-20 is capable to reduce the viral load by 2–3 log10 [49].
A disadvantage of T-20 is that is has to be given parenterally. A further limitation is that single amino acid mutations are sufficient for a significant loss of activity such as G36, V38, and Q40, and to a lesser extent N42, N43 and L44, L45. The advantage is that T-20 acts independently of all enzymes and thus can be applied to patients with RT and PR resistant HIV, and that it has a low spectrum of side effects besides injection site reactions [49].
With the synthesis of T-20, it was shown that a theoretical principle after verification in tissue culture led to a drug applicable in humans, which might be used for other viruses as well [35, 49].
Viral entry—CCR5 coreceptor antagonists
The chemokine receptors 4 (CXCR4) and 5 (CCR5) are coreceptors for HIV after gp120 has bound to the CD4 molecule. A lymphotropic or syncytium inducing HIV will bind to CXCR4 (so called X4 virus), and a macrophagotropic or non-syncytium inducing virus to CCR5 (R5). Additionally, some viral strains can use both receptors (dual tropic). By analysis of the CCR5-molecule, it was found that in white Caucasians approximately 1% have a homozygous deletion in their genome (delta 32) resulting in the absence of the 7 membrane spanning G-protein on the surface of lymphocytes. The general state of health of these carriers was unaffected, but they were less susceptible to acquisition of R5-virus transmitted by sexual contact [4].
One of the substances developed to interfere with binding of HIV to the CCR5 coreceptor has been approved for ART at the end of 2007 (maraviroc), one further (vicirviroc) is pending (Table 1). In clinical trials, the evaluation of the antiviral efficacy of maraviroc showed a 2.5 log10 viral load reduction in short term monotherapy [50]. During treatment, it became evident that in most cases a shift of the viral tropism from R5 to X4 led to maraviroc failure by selecting a few mutations within the V3-loop of HIV-1; typical amino acid changes are acidic to neutral or neutral to basic [35, 50, 51]. These X4 variants were mainly present as minority species; however, in some cases selection of initially undetectable X4 strains has been observed [52, 53]. In addition, therapy failure without a tropism shift has been reported in some patients due to the selection of point mutations within the V3-loop causing resistance to maraviroc instead of leading to a tropism shift [54].
As mentioned above, the use of CCR5 inhibitors is limited to R5 viruses of the HIV-1 group M, and will not inhibit binding of HIV-2. During the natural course of HIV infection, X4 viruses finally dominate in AIDS patients, thus CCR5 inhibitors should be used earlier in the course of the disease. A phenotypic assay is generally used to determine the coreceptor binding, and genotypic approaches are currently evaluated. Until now, this unique strategy for HIV growth inhibition has not been extended for further viruses.
Maturation inhibitors
Cleavage of the gag/pol precursor protein of HIV-1 results in the release of the core protein. The enclosed capsid protein initially has a molecular size of 25.000 (p25), which is further cleaved into a smaller fragment (p24). This step can be inhibited by bevirimat, so that p25 will not be cleaved; in consequence the polymerization of p24 is prevented and the conic core is not formed [55, 56]. As learned from the mode of action of protease inhibitors, HIV released from cells after treatment with bevirimat will not be infectious [35].
Conclusions
While the knowledge of inhibition of some enzymes has been transferred from HIV to HBV and HCV, the blockage of adhesion of a virus by changing access to its receptor is still an open field to be applied for other viruses. In the case of HIV, it has been shown that inhibition of this step is effective and beneficial for the clinical outcome of diseased patients. Application of this strategy for further viruses is pending. In summary, the knowledge gained from the diagnostics and treatment of HIV has fertilized the field of clinically important viruses causing chronic disease such as HBV and HCV, and of epidemiologically important viruses such as influenza and Dengue fever virus, while the interaction of interferons [57] is important for HCV [58], but has never touched HIV basically and the area of small interfering RNA (si-RNA) is still a field to be evolved.
References
Gottlieb MD, Schroff R, Schanker HM, Weissman JD, Fan PT, Wolf RA, Saxon A (1981) Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men. N Engl J Med 305:1425–1431
Barré-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vézinet-Brun F, Rouzioux C, Rozenbaum W, Montagnier L (1983) Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220:868–871. doi:10.1126/science.6189183
Gallo RC, Sarin PS, Gelmann EP, Robert-Guroff M, Richardson E, Kalyanaraman VS, Mann D, Sidhu GD, Stahl RE, Zolla-Pazner S, Leibowitch J, Popovic M (1983) Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science 220:865–867. doi:10.1126/science.6601823
Levy JA (2007) HIV and the pathogensis of AIDS, 3rd edn. ASM Press, Washington DC
Gordon S, Pandrea I, Dunham R, Apretei C, Silvestri G (2005) The call of the wild: what can be learned from studies of SIV infection in natural hosts. HIV Seq Compendium 2005:2–29. Los Alamos, New Mexico
Barnett SW, Murthy KK, Herndier BG, Levy JA (1994) An AIDS like condition induced in baboons by HIV-2. Science 266:642–646. doi:10.1126/science.7939718
Onaga R, Kornfeld C, Pandrea I, Estaquier J, Souquière S, Rouquet P, Apetrei C, Poques P, Müller-Trutwin MC (2002) High levels of viral replication contrast with only transient changes in CD4+ and CD8+ cell numbers during the early phase of experimental infection with simian immunodeficiency virus SIVmnd-1 in Mandrillus sphinx. J Virol 76:10256–10263. doi:10.1128/JVI.76.20.10256-10263.2002
Van Heuerswyn F, Li Y, Neel C, Bailess E, Keele BF, Liu W, Loul S, Butel C, Liegeois F, Bienvenue Y, Ngolle EM, Sharp PM, Shaw GM, Delaporte E, Hahn BH, Peeters M (2006) SIV infection in wild gorillas. Nature 444:164. doi:10.1038/444164a
Fleming AP (1988) Seroepidemiology of human immunodeficiency viruses in Africa. Biomed Pharmacother 42:309–320
Worobey M, Gemmel M, Teuwen DE, Haselkorn T, Kunstman K, Bunce M, Muyembe JJ, Kabongo JMM, Kalengayi RM, Van Marck E, Gilbert MT, Wolinsky SM (2008) Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature 455:661–664. doi:10.1038/nature07390
Ndumbe PM, Atchou G, Biwole M, Lobe V, Ayuk-Takem J (1993) Infections among pygmies in the Eastern province of Cameroon. Med Microbiol Immunol (Berl) 182:281–284. doi:10.1007/BF00191943
Sharp PM, Hahn BH (2008) Prehistory of HIV-1. Nature 455:605–606. doi:10.1038/455605a
Leitner T, Korber B, Daniels M, Calef C, Foley B (2005) HIV-1 subtype and circulating recombinant form (CRF), references sequences, 2005. HIV Seq Compendium 2005:41–48. Los Alamos, New Mexico
Hemelaar J, Gouws E, Ghys PD, Osmanov S (2006) Global and regional distribution of HIV-1 genetic subtypes in 2004. AIDS 20:2011–2019. doi:10.1097/01.aids.0000247564.73009.bc
Courgnaud V, Abela B, Pourrut X, Mpoudi-Ngolle E, Loul S, Delaporte E, Peeters M (2003) Identification of a new simiam immunodeficiency lineage with a vpu gene present among different cercopithecus monkeys (C. mona, C. cephus, and C. nictitans) from Cameroon. J Virol 77:12523–12534. doi:10.1128/JVI.77.23.12523-12534.2003
Leitner T, Dazza MC, Ekwalanga M, Apetrei C, Saragosti S (2007) Sequence diversity among chimpanzee immunodeficiency viruses (SIVcpz) suggests that SIVcpzPts was derived from SIVcpzPtt through additional recombination events. AIDS Res Hum Retroviruses 23:1114–1118. doi:10.1089/aid.2007.0071
Nerrienet E, Santiago ML, Foupouapuognini Y, Bailes E, Mundy NI, Njinku B, Kfutwah A, Muller-Trutwin MC, Barre-Sinoussi F, Shaw GM, Sharp PM, Hahn BH, Ayouba A (2005) Simian immunodeficiency virus infection in wild caught chimpanzees from Cameroon. J Virol 79:1312–1319. doi:10.1128/JVI.79.2.1312-1319.2005
Van Heuverswyn F, Yingying LI, Bailes E, Neel C, Lafay B, Keele BF, Shaw KS, Takehisa J, Kraus MH, Loul S, Butel C, Liegeois F, Yangda B, Sharp PM, Mpoudi-Ngolle E, Delaporte E, Hahn BH, Peeters M (2007) Genetic diversity and phylogeographic clustering of SIVcpzPtt in wild chimpanzees in Cameroon. Virology 368:155–171. doi:10.1016/j.virol.2007.06.018
Quinn T (2008) HIV epidemiology and the effects of antiviral therapy on long-term consequences. AIDS 22(Suppl 3):S7–S12. doi:10.1097/01.aids.0000327510.68503.e8
Wendler I, Jentsch KD, Schneider J, Hunsmann G (1987) Efficient replication of HTLV-III and STLV-III mac in human Jurkat cells. Med Microbiol Immunol (Berl) 176:273–280. doi:10.1007/BF00190533
Nakashima H, Koyanagi Y, Harada S, Yamamoto N (1986) Effect of HTLV-III on the macromolecular synthesis in HTLV-I carrying cell line MT-4. Med Microbiol Immunol (Berl) 175:325–334. doi:10.1007/BF02123869
Owen SM, Yang C, Spira T, Ou CY, Pau CP, Parekh BS, Candal D, Kuehl D, Kennedy MS, Rudolph D, Low W, Delatorre N, Masciotra S, Kalish ML, Cowart F, Barnett T, Lal R, McDougal JS (2008) Alternative algorithm for human immunodeficiency virus infection diagnosis using tests that are licensed in United States. J Clin Microbiol 46:1588–1595. doi:10.1128/JCM.02196-07
Ayoagi K, Ohue C, Iida K, Kimura T, Tanaka E, Kiyosawa K, Yagi S (1999) Development of a single and highly sensitive enzyme immunoassay for hepatitis C virus core antigen. J Clin Microbiol 37:1802–1808
Veillon P, Payan C, Picchio G, Montreuil MM, Guntz P, Lunel F (2003) Comparative evaluation of the total hepatitis C virus core antigen, branched-DNA, and Amplicor Monitor assay in determining viremia for patients with chronic hepatitis C during interferon plus ribavirin combination therapy. J Clin Microbiol 41:3212–3220. doi:10.1128/JCM.41.7.3212-3220.2003
Rouet F, Rouzioux C (2007) The measurement of HIV-1 viral load in resource limited settings: how and where? Clin Lab (Zaragoza) 53:135–148
Schutten M, Peters D, Back NK, Beuselinck K, Foulongne V, Gerreti AM, Pandiani L, Tiemann C, Niesters HG (2007) Multicenter evaluation of the new Abbott Real Time assays for quantitative detection of human immunodeficiency virus type 1 and hepatitis C virus RNA. J Clin Microbiol 45:1712–1717. doi:10.1128/JCM.02385-06
Lefrere JJ, Coste J, Defer C, Mercier B, Loisseau P, Vignon D, Pawlotsky JM, Biagini P, Lerable J, Rouger P, Roudot-Thoraval F, Ferec C (1999) Screening for HBV, HCV and HIV genomes in blood donations: shortcomings of pooling revealed by a multicentre study simulating real-time testing. J Virol Methods 80:33–44. doi:10.1016/S0166-0934(99)00028-2
Offergeld R, Faensen D, Ritter S, Hamouda O (2005) Human immunodeficiency virus, hepatitis C and B infections among blood donors in Germany 2000–2002: risk of virus transmission and the impact of nucleic acid amplification testing. Euro Surveill 10:8–11
Hirsch MS, Günthard HF, Schapiro JM, Brun-Vézinet F, Clotet B, Hammer SM, Johnson VA, Kunitzkes DR, Mellors JW, Pillay D, Yeni PG, Jacobsen DM, Richman DD (2008) Antiretroviral drug resistance testing in adult HIV-1 infection: 2008 recommendation of the International AIDS society—USA panel. Clin Infect Dis 47:266–285. doi:10.1086/589297
Arens M (2001) Clinically relevant sequence-based genotyping of HBV, HCV, CMV and HIV. J Clin Virol 22:11–29. doi:10.1016/S1386-6532(01)00156-1
Klug SJ, Molijn A, Schopp B, Holz B, Iftner A, Quint WJF, Snijders P, Petry KU, Krueger Kjaer S, Munk C, Iftner T (2008) Comparison of the performance of different HPV genotyping methods for detection genital HPV types. J Med Virol 80:1264–1274. doi:10.1002/jmv.21191
Cinatl J, Michaelis M, Doerr HW (2007) The threat of avian influenza (H5N1). Part III: antiviral therapy. Med Microbiol Immunol (Berl) 196:203–212. doi:10.1007/s00430-007-0048-z
Plantier JC, Dachraoui R, Lemee V, Guedin M, Borsa-Lebas F, Caron F, Simon F (2005) HIV-1 resistance genotyping on dried serum spots. AIDS 19:391–397. doi:10.1097/01.aids.0000161768.98534.e7
Ho M (2008) The history of cytomegalovirus and its disease. Med Microbiol Immunol (Berl) 197:65–73. doi:10.1007/s00430-007-0066-x
DeClercq E (2007) The design of drugs for HIV and HCV. Nat Rev Drug Discov 12:1001–1018. doi:10.1038/nrd2424
Sloan RD, Ijaz S, Moore PL, Harrison TJ, Teo CG, Tedder RS (2008) Antiviral resistance mutations potentiate hepatitis B virus immune evasion through disruption of its surface antigen a determinant. Antivir Ther 13:439–447
Menne B, Butter SD, George AL, Tochkov IA, Zhu Y, Xiong S, Gerin JL, Cote PJ, Tennant BC (2008) Antiviral effects of lamivudine, emtricitabine, adefovir dipivoxil, and tenofovir disoproxil fumarate administered orally alone and in combination to woodchucks with chronic woodchuck hepatitis virus infection. Antimicrob Agents Chemother 52:3617–3632. doi:10.1128/AAC.00654-08
Manns MP, Foster GR, Rockstroh JK, Zeuzem S, Zoulim F, Houghton M (2007) The way forward in HCV treatment—finding the right path. Nat Rev Drug Discov 12:991–1000. doi:10.1038/nrd2411
Brown TT (2008) Approach to the human immunodeficiency virus-infected patient with lipodystrophy. J Clin Endocrinol Metab 93:2937–2945. doi:10.1210/jc.2008-1019
Savoia D, Allice T, Tovo PA (2005) Antileishmanial activity of HIV protease inhibitors. Int J Antimicrob Agents 26:92–94. doi:10.1016/j.ijantimicag.2005.04.003
Trudel N, Garg R, Messier N, Sundar S, Ouellette M, Tremblay MJ (2008) Intracellular survival of Leishmania species that cause visceral leishmaniasis is significantly reduced by HIV-1 protease inhibitors. J Infect Dis 198:1292–1299. doi:10.1086/592280
He Y, King MS, Kempf DJ, Lu L, Lim HB, Krishnan P, Kati W, Middleton T, Molla A (2008) Relative replication capacity and selective advantage profiles of protease inhibitor-resistant hepatitis C virus (HCV) NS3 protease mutants in the HCV genotype 1b replicon system. Antimicrob Agents Chemother 52:1101–1110. doi:10.1128/AAC.01149-07
Forestier N, Reesink HW, Weegink CJ, McNair L, Kieffer TL, Chu HM, Purdy S, Jansen PL, Zeuzem S (2007) Antiviral activity of telaprevir (VX-950) and peginterferon-2α in patients with hepatitis C. Hepatology 46:640–648. doi:10.1002/hep.21774
Soriano V, Peters MG, Zeuzem S (2009) New therapies for hepatitis C virus infection. Clin Infect Dis 48:313–320. doi:10.1086/595848
Grinszteijn B, Nguyen BY, Katlama C, Gatell JM, Lazzarin A, Vittecoq D, Gonzalez CJ, Chen J, Harvey CM, Isaacs RD (2007) Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II randomised controlled trial. Lancet 369:1261–1269. doi:10.1016/S0140-6736(07)60597-2
Iwamoto M, Wenning LA, Petry AS, Laethem M, de Smet M, Kost JT, Breidinger SA, Mangin EC, Azrolan N, Greenberg HE, Haazen W, Stone JA, Gottesdiener KM, Wagner JA (2008) Minimal effects of ritonavir and efavirenz on the pharmacogenetic of raltegravir. Antimicrob Agents Chemother 52:4338–4343. doi:10.1128/AAC.01543-07
Hombrouck A, Van Remoortel B, Michiels M, Noppe W, Christ F, Eneroth A, Sahlberg BL, Benkestock K, Vrang L, Johansson NG, Barecca ML, De Luca L, Ferro S, Chimirri A, Debyser Z, Witvrouw M (2008) Preclinical evaluation of 1H-benzylindole derivatives as novel human immunodeficiency virus integrase strand transfer inhibitors. Antimicrob Agents Chemother 52:2861–2869. doi:10.1128/AAC.00210-08
Malet I, Delelis O, Valentin MA, Montes B, Soulie C, Wirden M, Tchertanov L, Peytavin G, Reynes J, Mouscadet JF, Katlama C, Calvez V, Marcelin AG (2008) Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob Agents Chemother 52:1351–1358. doi:10.1128/AAC.01228-07
Morozov VA, Morozov AV, Schürmann D, Jessen H, Kücherer C (2007) Transmembrane protein polymorphism and resistance to T-20 (Enfurtide, Fuzeon) in HIV-1 infected therapy-naive seroconverters and AIDS patients under HAART-T-20 therapy. Virus Genes 35:167–174. doi:10.1007/s11262-007-0098-8
Mac Arthur RD, Novak RM (2008) Reviews of anti-infective agents: maraviroc: the first of a new class of antiretroviral agents. Clin Infect Dis 47:236–241. doi:10.1086/589289
Briggs DR, Tuttle DL, Sleasman JW, Goodenow MM (2000) Envelope V3 amino acid sequence predicts HIV-1 phenotype (co-receptor usage and tropism for macrophages). AIDS 14:2937–2939. doi:10.1097/00002030-200012220-00016
Lewis M, Simpson P, Fransen S, Huang W, Whitcomb J, Mosley M, Robertson DL, Mansfield R, Ciaramella G, Westby M (2007) CXCR4-using virus detected in patients receiving maraviroc in the Phase III studies MOTIVATE 1 and 2 originates from a pre-existing minority of CXCR4-using virus. Antivir Ther 12:S65
Gulick RM, Lalezari J, Goodrich J, Clumeck N, De Jesus E, Horban A, Nadler J, Clotet B, Karlsson A, Wohlfeiler M, Montana JB, McHale M, Sullivan J, Ridgway C, Felstead S, Dunne MW, van der Ryst E, Mayer H, MOTIVATE study teams (2008) Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med 359:1429–1441. doi:10.1056/NEJMoa0803152
Lewis M, Simpson P, Whitcomb J, Li X, Robertson DL, Westby M (2008) Changes in V3 loop sequence associated with failure of maraviroc treatment in patients enrolled in the MOTIVATE 1 and 2 trials. In: 15th conference on retroviruses and opportunistic infections. Boston, 2008. Poster 871
Van Baelen K, Salzwedel K, Rondelez E, Van Eygen V, De Vos S, Verheyen A, Steegen K, Verlinden Y, Allaway GP, Stuyver LJ (2009) Susceptibility of human immunodeficiency virus type 1 to the maturation inhibitor Bevirimat is modulated by baseline polymorphisms in gag spacer peptide 1. Antimicrob Agents Chemother 53:2185–2188. doi:10.1128/AAC.01650-08
Adamson CS, Waki K, Ablan SD, Salzwedel K, Freed EO (2009) Impact of human immunodeficiencyvirus type 1 resistance to protease inhibitors on evolution of resistance to the maturation inhibitor Bevirimat (PA-457). J Virol 83:4884–4894. doi:10.1128/JVI.02659-08
Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR (2007) Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 12:975–990. doi:10.1038/nrd2422
Goyal A, Hofmann WP, Hermann E, Traver S, Hissar SS, Aroya N, Blum HE, Zeuzem S, Sarrazin C, Sarin SK (2007) The hepatitis C virus NS5A protein and response to interferon α: mutational analyses in patients with chronic HCV genotype 3a infection from India. Med Microbiol Immunol (Berl) 196:11–21. doi:10.1007/s00430-006-0024-z
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Stürmer, M., Doerr, H.W. & Gürtler, L. Human immunodeficiency virus: 25 years of diagnostic and therapeutic strategies and their impact on hepatitis B and C virus. Med Microbiol Immunol 198, 147–155 (2009). https://doi.org/10.1007/s00430-009-0117-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00430-009-0117-6