Key Players of Cisplatin Resistance: Towards a Systems Pharmacology Approach

 , , and
, , and
Abstract
:
1. Introduction
2. Results
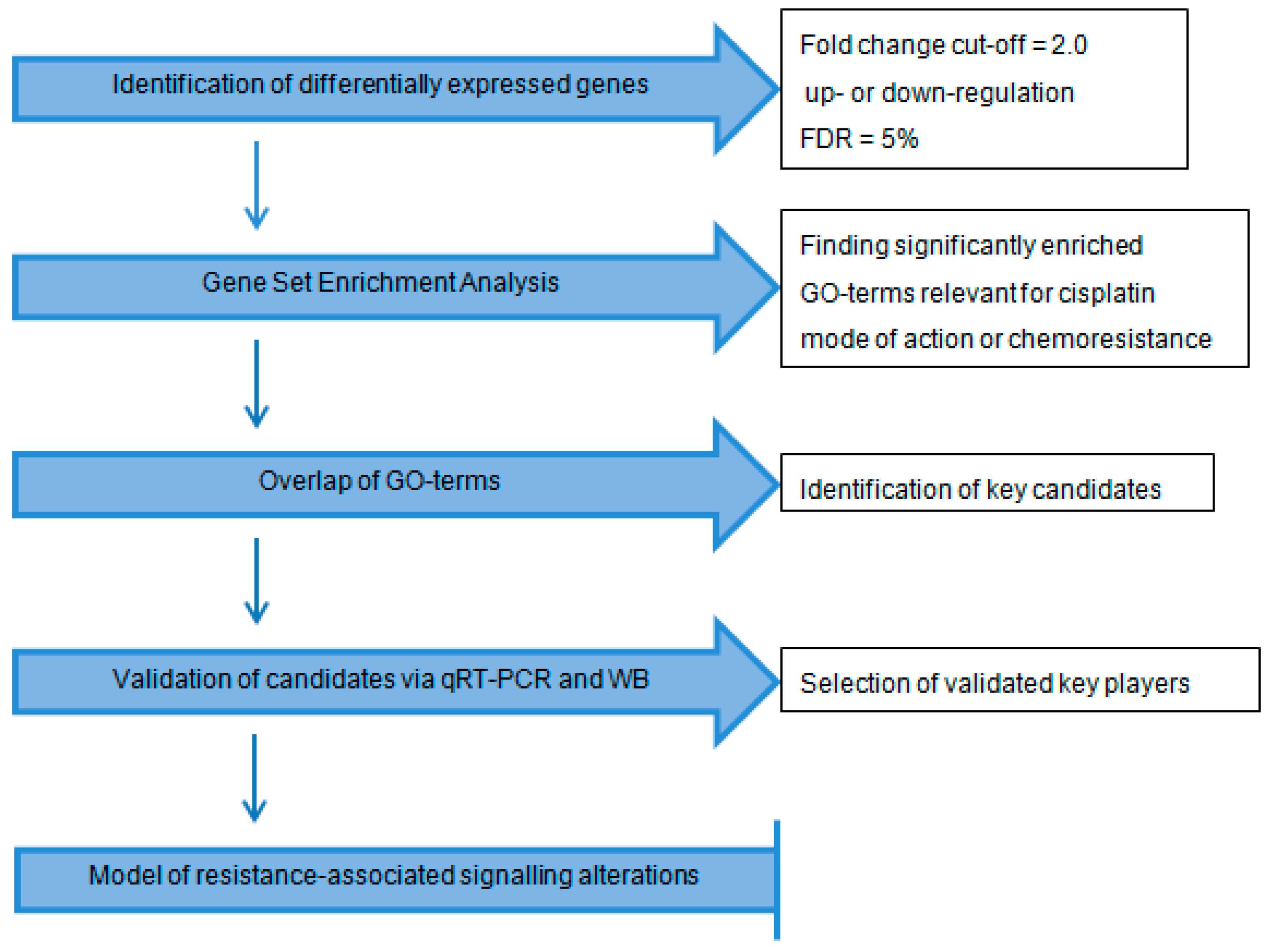
2.1. Workflow
2.2. Microarray Analysis
2.2.1. Differentially Expressed Genes
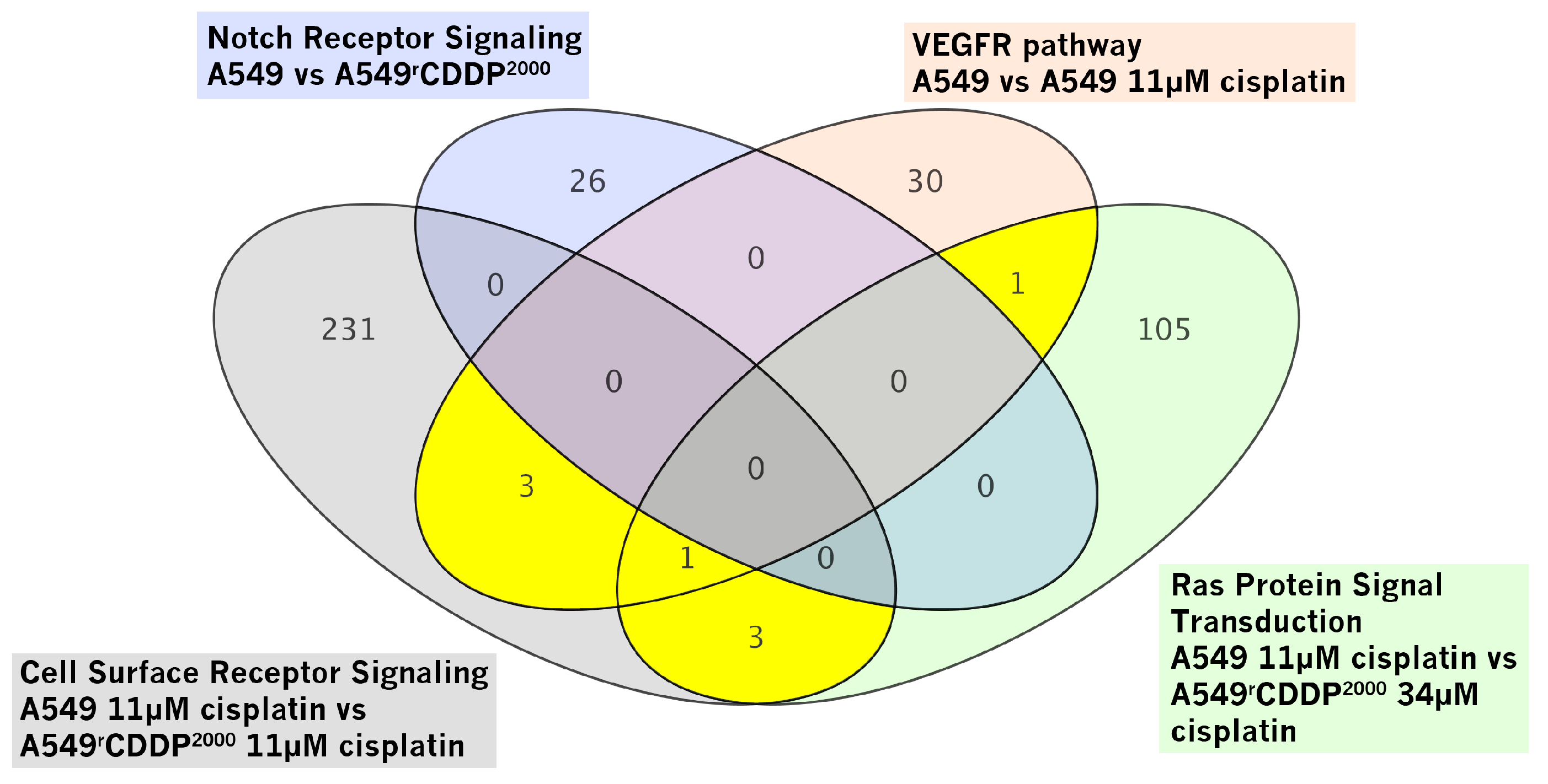
2.2.2. Gene Set Enrichment Analysis
2.3. Evaluation of the Identified Candidates
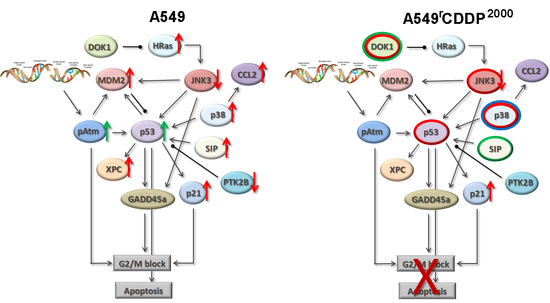
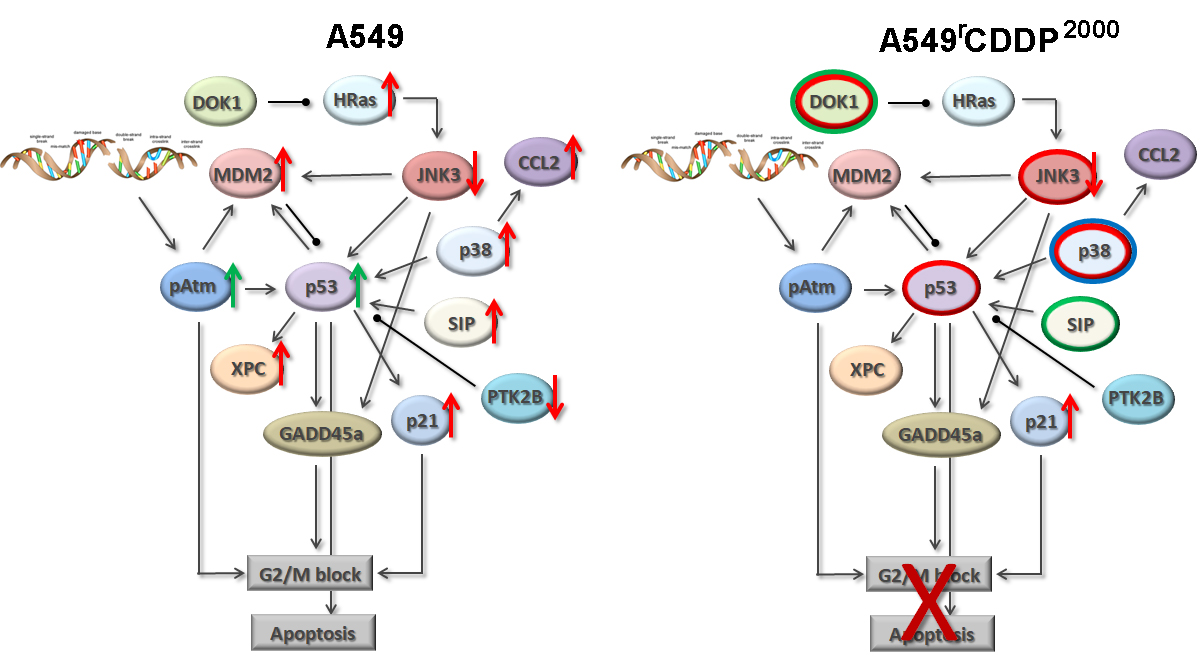
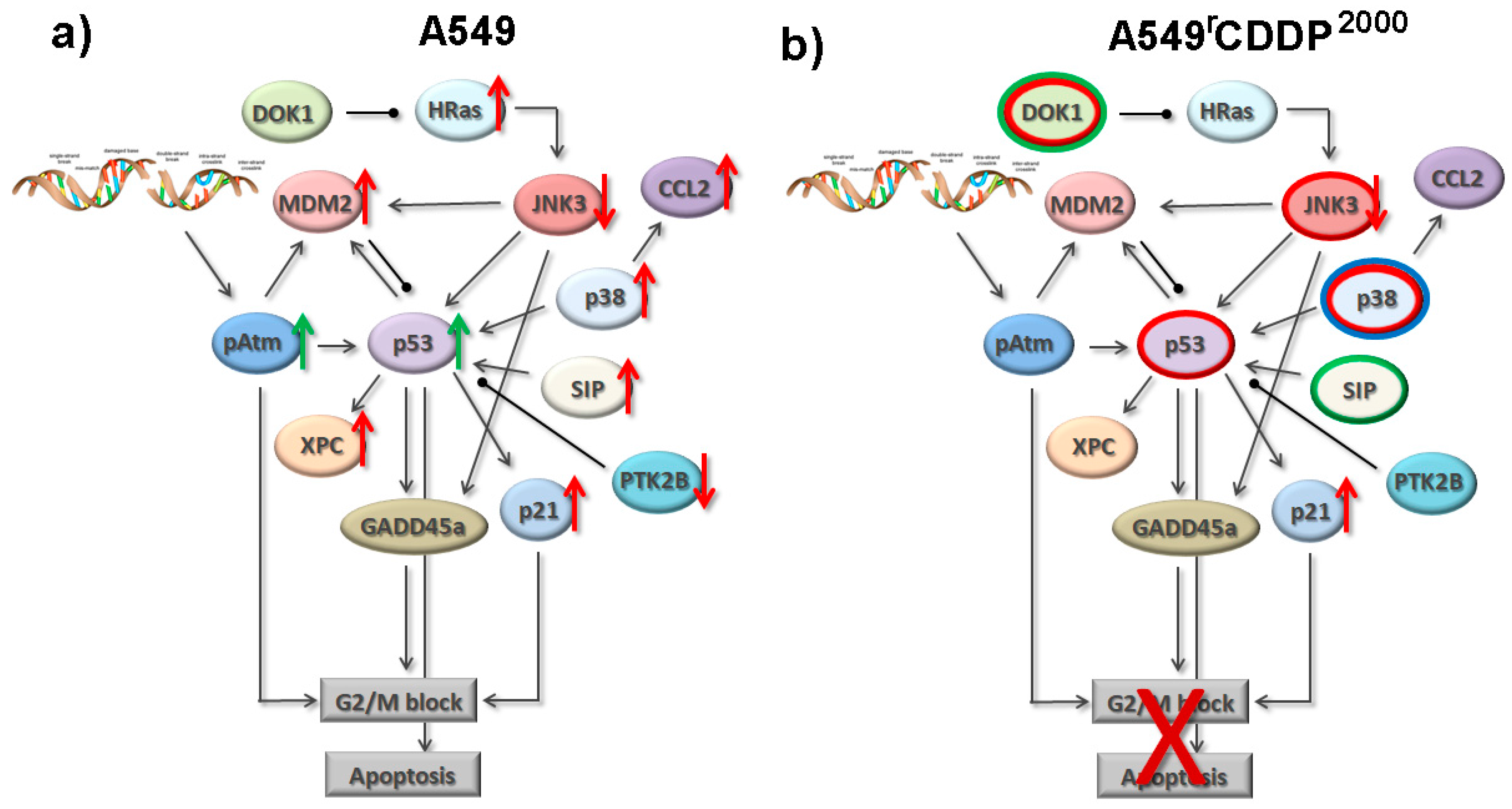
2.4. Extended Model of Resistance-Associated Signalling Alterations
3. Discussion
3.1. Systems Pharmacology Approach
3.2. Role of the Identified Key Players
3.3. Model of Resistance-Associated Signalling Alterations
4. Materials and Methods
4.1. Drugs
4.2. Cell Lines
4.3. Microarray
4.4. RNA Isolation, cDNA Synthesis and qRT-PCR
4.5. SDS-PAGE and Western Blot
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| NSCLC | non-small cell lung cancer |
| CDDP, DDP | cis-diamminedichloroplatinum (II) (cisplatin) |
| GO | Gene Ontology |
| GSEA | Gene Set Enrichment Analysis |
| FDR | false discovery rate |
| VEGFR | vascular endothelial growth factor receptor |
| TBS | Tris-buffered saline |
| MAPK | mitogen-activated protein kinase |
| ERK | Extracellular-signal Regulated Kinase |
| DOK1 | Docking protein 1 |
| PTK2B, Pyk2 | Protein tyrosine kinase 2 beta |
| CCL2 | C-C motif chemokine ligand 2 |
| MCP-1 | monocyte chemotactic protein 1 |
| MAPKAPK2 | MAP kinase-activated protein kinase 2 |
| JNK | c-Jun N-terminal kinase |
| MDM2 | mouse double minute 2 homolog |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
References
- Besse, B.; Adjei, A.; Baas, P.; Meldgaard, P.; Nicolson, M.; Paz-Ares, L.; Reck, M.; Smit, E.F.; Syrigos, K.; Stahel, R.; et al. 2nd ESMO Consensus Conference on Lung Cancer: Non-small-cell lung cancer first-line/second and further lines of treatment in advanced disease. Ann. Oncol. 2014, 25, 1475–1484. [Google Scholar] [CrossRef] [PubMed]
- Shtivelman, E.; Hensing, T.; Simon, G.R.; Dennis, P.A.; Otterson, G.A.; Bueno, R.; Salgia, R. Molecular pathways and therapeutic targets in lung cancer. Oncotarget 2014, 5, 1392–1433. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, Z.M.; Li, X.C.; Yao, Y.T.; Yin, Z.X. Activation of ERK1/2 and Akt is associated with cisplatin resistance in human lung cancer cells. J. Chemother. 2013, 25, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Jordan, P.; Carmo-Fonseca, M. Molecular mechanisms involved in cisplatin cytotoxicity. Cell. Mol. Life Sci. 2000, 57, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Fajac, A.; Da Silva, J.; Ahomadegbe, J.C.; Rateau, J.G.; Bernaudin, J.F.; Riou, G.; Bénard, J. Cisplatin-induced apoptosis and p53 gene status in a cisplatin-resistant human ovarian carcinoma cell line. Int. J. Cancer 1996, 68, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Sarin, N.; Engel, F.; Kalayda, G.V.; Mannewitz, M.; Cinatl, J.; Rothweiler, F.; Michaelis, M.; Saafan, H.; Ritter, C.A.; Jaehde, U.; et al. Cisplatin resistance in non-small cell lung cancer cells is associated with an abrogation of cisplatin-induced G2/M cell cycle arrest. PLoS ONE 2017, 12, e0181081. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Senovilla, L.; Eisenberg, T.; Carmona-Gutierrez, D.; Vacchelli, E.; Robert, T.; Ripoche, H.; Jägemann, N.; Paccard, C.; et al. Independent transcriptional reprogramming and apoptosis induction by cisplatin. Cell Cycle 2012, 11, 3472–3480. [Google Scholar] [CrossRef] [PubMed]
- Toshimitsu, H.; Hashimoto, K.; Tangoku, A.; Iizuka, N.; Yamamoto, K.; Kawauchi, S.; Oga, A.; Furuya, T.; Oka, M.; Sasaki, K. Molecular signature linked to acquired resistance to cisplatin in oesophageal cancer cells. Cancer Lett. 2004, 211, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Gatti, L.; Chen, D.; Beretta, G.L.; Rustici, G.; Carenini, N.; Corna, E.; Colangelo, D.; Zunino, F.; Bähler, J.; Perego, P. Global gene expression of fission yeast in response to cisplatin. Cell. Mol. Life Sci. 2004, 61, 2253–2263. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.C.; Manorek, G.; Samimi, G.; Lin, X.; Berry, C.C.; Howell, S.B. Identification of genes whose expression is associated with cisplatin resistance in human ovarian carcinopma cells. Cancer Chemother. Pharmacol. 2006, 58, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, H.; Hou, S.; Hu, B.; Liu, J.; Wang, J. Differences in gene expression profiles and carcinogenesis pathways involved in cisplatin resistance of four types of cancer. Oncol. Rep. 2013, 30, 596–614. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, H.; Hou, S.; Hu, B.; Liu, J.; Wang, J. The noncoding RNA expression profile and the effect of lncRNA AK126698 on cisplatin resistance in non-small-cell lung cancer cell. PLoS ONE 2013, 8, e65309. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Chen, J.; Zhang, F.; Wang, J.; Pan, J.; Chen, J.; Wang, Y. Aberrant Long Noncoding RNAs Expression Profiles Affect Cisplatin Resistance in Lung Adenocarcinoma. BioMed Res. Int. 2017, 2017, 7498151. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhang, C.; Wu, T.; Wang, Q.; Liu, J.; Dai, P. Transcriptome Sequencing Reveals Key Pathways and Genes Associated with Cisplatin Resistance in Lung Adenocarcinoma A549 Cells. PLoS ONE 2017, 12, e0170609. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.; White, J.T.; Yan, X.; Collins, S.; Drescher, C.W.; Urban, N.D.; Hood, L.; Lin, B. Proteins associated with Cisplatin resistance in ovarian cancer cells identified by quantitative proteomic technology and integrated with mRNA expression levels. Mol. Cell. Proteom. 2006, 5, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.-Z.; Qu, Y.-Q.; Zhang, W.-J.; Xiu, B.; Deng, A.-M.; Liang, A.-B. Proteomic analysis identified DJ-1 as a cisplatin resistant marker in non-small cell lung cancer. Int. J. Mol. Sci. 2011, 12, 3489–3499. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.L.; Madian, A.G.; Williams, S.W.; Chen, V.; Wing, C.; Hause, R.J.; To, L.A.; Gill, A.L.; Myers, J.L.; Gorsic, L.K.; et al. Identification of Novel Protein Expression Changes Following Cisplatin Treatment and Application to Combination Therapy. J. Proteome Res. 2017, 16, 4227–4236. [Google Scholar] [CrossRef] [PubMed]
- Wist, A.D.; Berger, S.I.; Iyengar, R. Systems pharmacology and genome medicine: A future perspective. Genome Med. 2009, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Goeman, J.J.; van de Geer, S.A.; de Kort, F.; van Houwelingen, H.C. A global test for groups of genes: Testing association with a clinical outcome. Bioinformatics 2004, 20, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Terfve, C.; Rose, J.C.; Markowetz, F. HTSanalyzeR: An R/Bioconductor package for integrated network analysis of high-throughput screens. Bioinformatics 2011, 27, 879–880. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Mao, Z.; Huang, J.; Xie, S.; Liu, T.; Mao, Z. Blocking the NOTCH pathway can inhibit the growth of CD133-positive A549 cells and sensitize to chemotherapy. Biochem. Biophys. Res. Commun. 2014, 444, 670–675. [Google Scholar] [CrossRef] [PubMed]
- McAuliffe, S.M.; Morgan, S.L.; Wyant, G.A.; Tran, L.T.; Muto, K.W.; Chen, Y.S.; Chin, K.T.; Partridge, J.C.; Poole, B.B.; Cheng, K.-H.; et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E2939–E2948. [Google Scholar] [CrossRef] [PubMed]
- Daenen, L.G.M.; Roodhart, J.M.L.; van Amersfoort, M.; Dehnad, M.; Roessingh, W.; Ulfman, L.H.; Derksen, P.W.B.; Voest, E.E. Chemotherapy enhances metastasis formation via VEGFR-1-expressing endothelial cells. Cancer Res. 2011, 71, 6976–6985. [Google Scholar] [CrossRef] [PubMed]
- Sini, P.; Samarzija, I.; Baffert, F.; Littlewood-Evans, A.; Schnell, C.; Theuer, A.; Christian, S.; Boos, A.; Hess-Stumpp, H.; Foekens, J.A.; et al. Inhibition of multiple vascular endothelial growth factor receptors (VEGFR) blocks lymph node metastases but inhibition of VEGFR-2 is sufficient to sensitize tumor cells to platinum-based chemotherapeutics. Cancer Res. 2008, 68, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Granados, M.L.; Hudson, L.G.; Samudio-Ruiz, S.L. Contributions of the Epidermal Growth Factor Receptor to Acquisition of Platinum Resistance in Ovarian Cancer Cells. PLoS ONE 2015, 10, e0136893. [Google Scholar] [CrossRef] [PubMed]
- Juliachs, M.; Muñoz, C.; Moutinho, C.A.; Vidal, A.; Condom, E.; Esteller, M.; Graupera, M.; Casanovas, O.; Germà, J.R.; Villanueva, A.; et al. The PDGFRβ-AKT pathway contributes to CDDP-acquired resistance in testicular germ cell tumors. Clin. Cancer Res. 2014, 20, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Tsigelny, I.F.; Götz, A.W.; Howell, S.B. Cisplatin inhibits MEK1/2. Oncotarget 2015, 6, 23510–23522. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, Y.; Li, Y.; Zhou, J.; Wu, Y.; Cui, Y.; Yang, G.; Hong, Y. Mutations of p53 and KRAS activate NF-κB to promote chemoresistance and tumorigenesis via dysregulation of cell cycle and suppression of apoptosis in lung cancer cells. Cancer Lett. 2015, 357, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [PubMed]
- Rane, M.J.; Coxon, P.Y.; Powell, D.W.; Webster, R.; Klein, J.B.; Pierce, W.; Ping, P.; McLeish, K.R. p38 Kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J. Biol. Chem. 2001, 276, 3517–3523. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Gruol, D.L. The chemokine CCL2 activates p38 mitogen-activated protein kinase pathway in cultured rat hippocampal cells. J. Neuroimmunol. 2008, 199, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Warner, G.M.; Yin, P.; Knudsen, B.E.; Cheng, J.; Butters, K.A.; Lien, K.R.; Gray, C.E.; Garovic, V.D.; Lerman, L.O.; et al. Inhibition of p38 MAPK attenuates renal atrophy and fibrosis in a murine renal artery stenosis model. Am. J. Physiol. Ren. Physiol. 2013, 304, F938–F947. [Google Scholar] [CrossRef] [PubMed]
- Mercier, P.-L.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.-C.; Ghani, K.; Têtu, B.; Bairati, I.; Bachvarov, D. Characterization of DOK1, a candidate tumor suppressor gene, in epithelial ovarian cancer. Mol. Oncol. 2011, 5, 438–453. [Google Scholar] [CrossRef] [PubMed]
- Geng, W.; Ng, K.T.P.; Sun, C.K.W.; Yau, W.L.; Liu, X.B.; Cheng, Q.; Poon, R.T.P.; Lo, C.M.; Man, K.; Fan, S.T. The role of proline rich tyrosine kinase 2 (Pyk2) on cisplatin resistance in hepatocellular carcinoma. PLoS ONE 2011, 6, e27362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.-T.; Miller, N.L.G.; Nam, J.-O.; Chen, X.L.; Lim, Y.; Schlaepfer, D.D. Pyk2 inhibition of p53 as an adaptive and intrinsic mechanism facilitating cell proliferation and survival. J. Biol. Chem. 2010, 285, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Mashima, R.; Hishida, Y.; Tezuka, T.; Yamanashi, Y. The roles of Dok family adapters in immunoreceptor signaling. Immunol. Rev. 2009, 232, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Binétruy, B.; Smeal, T.; Karin, M. Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature 1991, 351, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Smeal, T.; Binetruy, B.; Mercola, D.A.; Birrer, M.; Karin, M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature 1991, 354, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.Y.; Adler, V.; Pincus, M.R.; Ronai, Z. MEKK1/JNK signaling stabilizes and activates p53. Proc. Natl. Acad. Sci. USA 1998, 95, 10541–10546. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.S. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol. Ther. 2004, 3, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Hildesheim, J.; Fornace, A.J. Gadd45a: An elusive yet attractive candidate gene in pancreatic cancer. Clin. Cancer Res. 2002, 8, 2475–2479. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Prieto, R.; Rojas, J.M.; Taya, Y.; Gutkind, J.S. A role for the p38 mitogen-acitvated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. 2000, 60, 2464–2472. [Google Scholar] [PubMed]
- Wang, X.; Martindale, J.L.; Holbrook, N.J. Requirement for ERK activation in cisplatin-induced apoptosis. J. Biol. Chem. 2000, 275, 39435–39443. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Senovilla, L.; Olaussen, K.A.; Pinna, G.; Eisenberg, T.; Goubar, A.; Martins, I.; Michels, J.; Kratassiouk, G.; et al. Prognostic impact of vitamin B6 metabolism in lung cancer. Cell Rep. 2012, 2, 257–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, T.; Guell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Arbiser, J.L.; Moses, M.A.; Fernandez, C.A.; Ghiso, N.; Cao, Y.; Klauber, N.; Frank, D.; Brownlee, M.; Flynn, E.; Parangi, S.; et al. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc. Natl. Acad. Sci. USA 1997, 94, 861–866. [Google Scholar] [CrossRef] [PubMed]
- To, M.D.; Wong, C.E.; Karnezis, A.N.; Del Rosario, R.; Di Lauro, R.; Balmain, A. Kras regulatory elements and exon 4A determine mutation specificity in lung cancer. Nat. Genet. 2008, 40, 1240–1244. [Google Scholar] [CrossRef] [PubMed]
- Sklar, M.D. Increased resistance to cis-diamminedichloroplatinum(II) in NIH 3T3 cells transformed by ras oncogenes. Cancer Res. 1988, 48, 793–797. [Google Scholar] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Atayar, C.; Rosati, S.; Bosga-Bouwer, A.; Kluin, P.; Visser, L. JNK is constitutively active in mantle cell lymphoma: Cell cycle deregulation and polyploidy by JNK inhibitor SP600125. J. Pathol. 2009, 218, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Niedner, H.; Christen, R.; Lin, X.; Kondo, A.; Howell, S.B. Identification of genes that mediate sensitivity to cisplatin. Mol. Pharmacol. 2001, 60, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.-M.; Chen, K.; Wang, Y.-C.; Lee, P.-J.; Wang, Y.-C. Elevated p53 and p21waf1 mRNA expression in blood lymphocytes from lung cancer patients with chemoresistance. Cancer Detect. Prev. 2007, 31, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Vega, G.G.; Avilés-Salas, A.; Chalapud, J.R.; Martinez-Paniagua, M.; Pelayo, R.; Mayani, H.; Hernandez-Pando, R.; Martinez-Maza, O.; Huerta-Yepez, S.; Bonavida, B.; et al. P38 MAPK expression and activation predicts failure of response to CHOP in patients with Diffuse Large B-Cell Lymphoma. BMC Cancer 2015, 15, 722. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Igea, A.; Canovas, B.; Dolado, I.; Nebreda, A.R. Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-induced apoptosis mediated by reactive oxygen species and JNK. EMBO Mol. Med. 2013, 5, 1759–1774. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-S.; Weng, S.-H.; Chen, H.-J.; Chiu, Y.-F.; Huang, Y.-C.; Tseng, S.-C.; Kuo, Y.-H.; Lin, Y.-W. Inhibition of p38 MAPK-dependent excision repair cross-complementing 1 expression decreases the DNA repair capacity to sensitize lung cancer cells to etoposide. Mol. Cancer Ther. 2012, 11, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Fader, A.N.; Rasool, N.; Vaziri, S.A.J.; Kozuki, T.; Faber, P.W.; Elson, P.; Biscotti, C.V.; Michener, C.M.; Rose, P.G.; Rojas-Espaillat, L.; et al. CCL2 expression in primary ovarian carcinoma is correlated with chemotherapy response and survival outcomes. Anticancer Res. 2010, 30, 4791–4798. [Google Scholar] [PubMed]
- Levina, V.; Su, Y.; Nolen, B.; Liu, X.; Gordin, Y.; Lee, M.; Lokshin, A.; Gorelik, E. Chemotherapeutic drugs and human tumor cells cytokine network. Int. J. Cancer 2008, 123, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Rothweiler, F.; Barth, S.; Cinatl, J.; van Rikxoort, M.; Löschmann, N.; Voges, Y.; Breitling, R.; von Deimling, A.; Rödel, F.; et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011, 2, e243. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Silver, J.; Oshlack, A.; Holmes, M.; Diyagama, D.; Holloway, A.; Smyth, G.K. A comparison of background correction methods for two-colour microarrays. Bioinformatics 2007, 23, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3. [Google Scholar] [CrossRef] [PubMed]



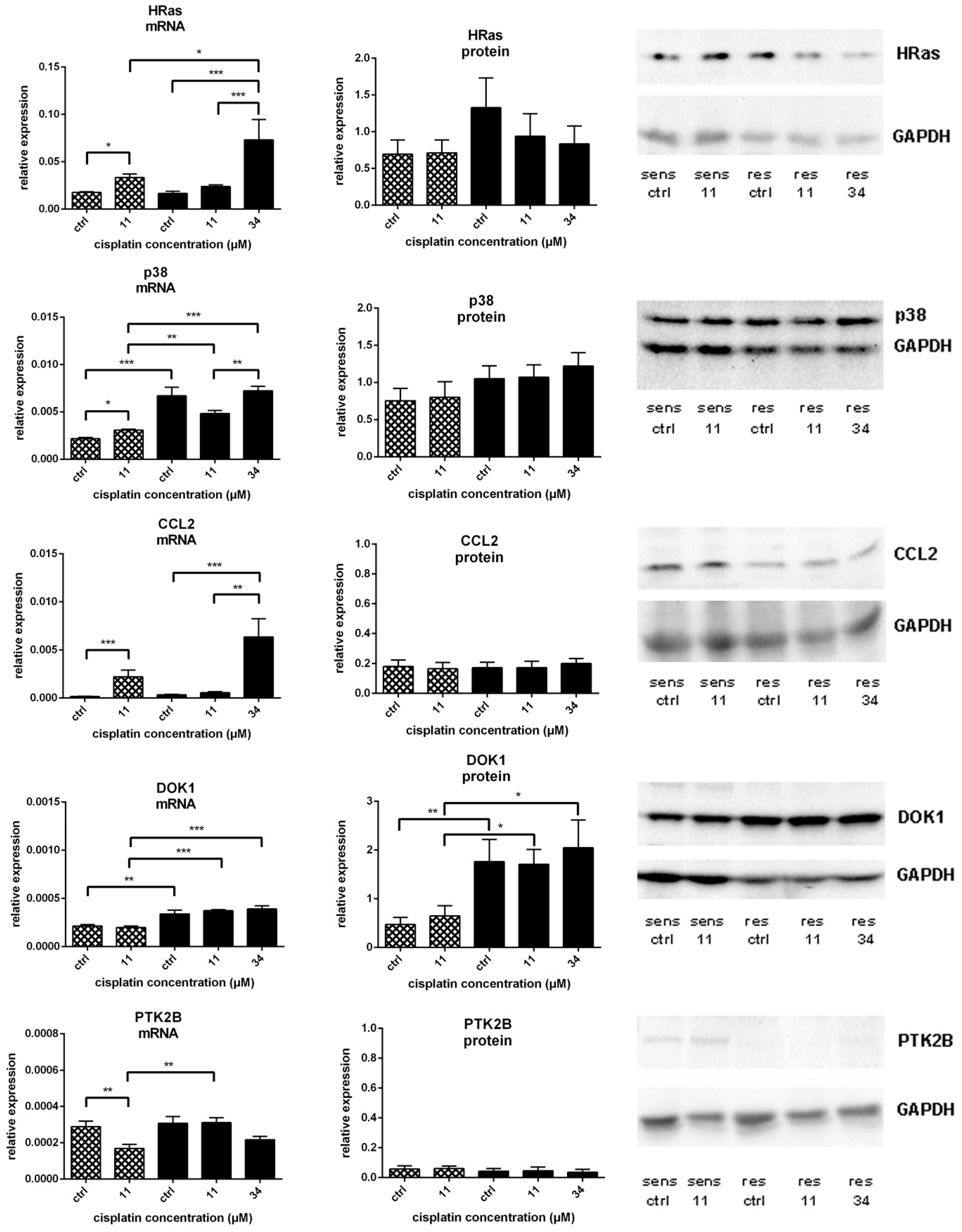
 ) and A549rCDDP2000 (
) and A549rCDDP2000 (  ) before (ctrl) and after treatment with 11 µM cisplatin (11) or 34 µM cisplatin (34) presented as mean ± SEM; as well as representative Western blots. * p < 0.05; ** p < 0.01; *** p < 0.01.
) and A549rCDDP2000 ( ) before (ctrl) and after treatment with 11 µM cisplatin (11) or 34 µM cisplatin (34) presented as mean ± SEM; as well as representative Western blots. * p < 0.05; ** p < 0.01; *** p < 0.01.
) before (ctrl) and after treatment with 11 µM cisplatin (11) or 34 µM cisplatin (34) presented as mean ± SEM; as well as representative Western blots. * p < 0.05; ** p < 0.01; *** p < 0.01.
) and A549rCDDP2000 ( ) before (ctrl) and after treatment with 11 µM cisplatin (11) or 34 µM cisplatin (34) presented as mean ± SEM; as well as representative Western blots. * p < 0.05; ** p < 0.01; *** p < 0.01.
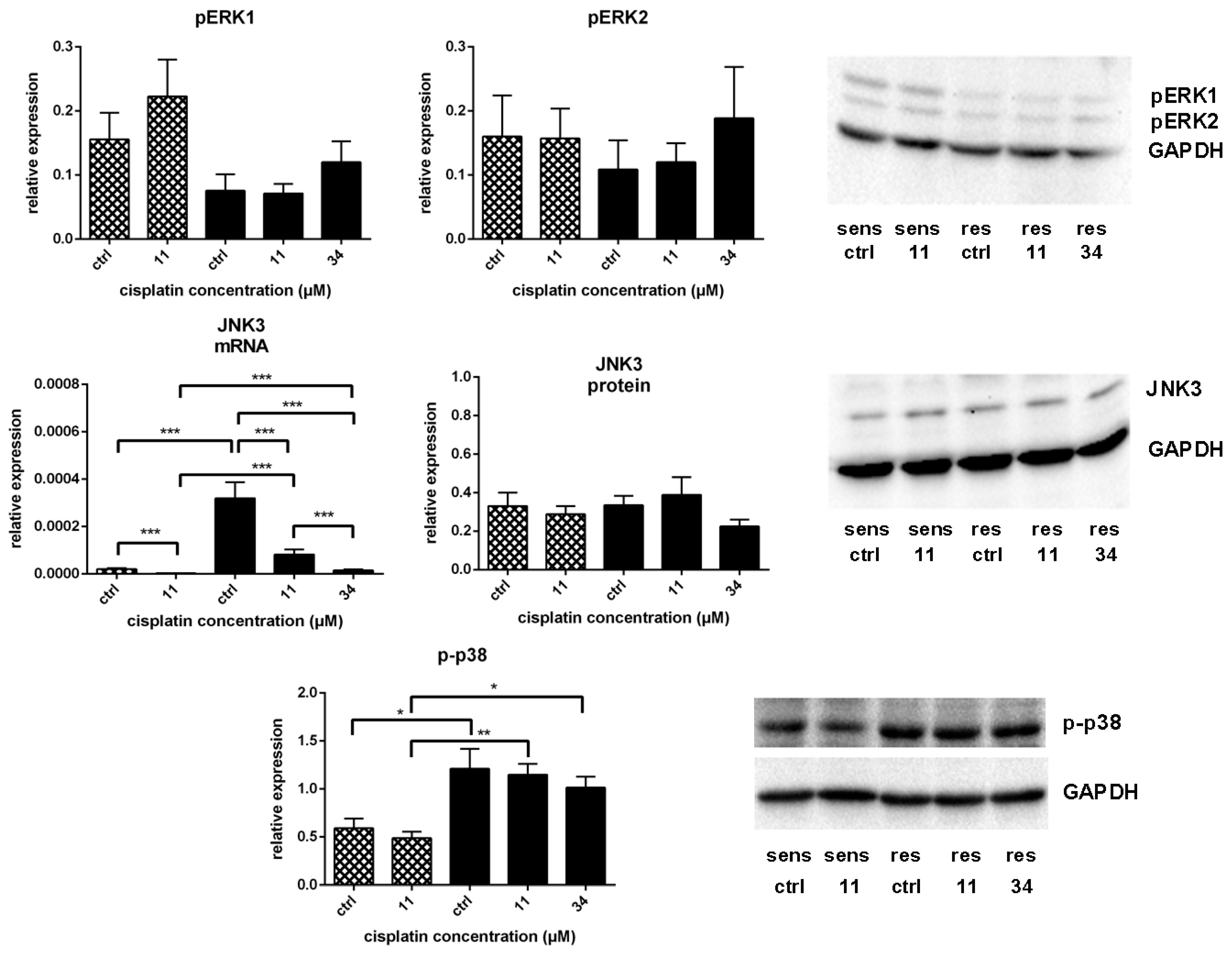
 ) and A549rCDDP2000 (
) and A549rCDDP2000 (  ) after treatment with 11 µM cisplatin (11) or 34 µM cisplatin (34) expressed as mean ± SEM, as well as representative Western blots.* p < 0.05; ** p < 0.01; *** p < 0.01.
) and A549rCDDP2000 ( ) after treatment with 11 µM cisplatin (11) or 34 µM cisplatin (34) expressed as mean ± SEM, as well as representative Western blots.* p < 0.05; ** p < 0.01; *** p < 0.01.
) after treatment with 11 µM cisplatin (11) or 34 µM cisplatin (34) expressed as mean ± SEM, as well as representative Western blots.* p < 0.05; ** p < 0.01; *** p < 0.01.
) and A549rCDDP2000 ( ) after treatment with 11 µM cisplatin (11) or 34 µM cisplatin (34) expressed as mean ± SEM, as well as representative Western blots.* p < 0.05; ** p < 0.01; *** p < 0.01.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Condition 1 | Treatment Condition 2 | Number of Differentially Expressed Genes |
|---|---|---|
| A549, untreated | A549rCDDP2000, untreated | 3697 |
| A549, 11 µM cisplatin | A549rCDDP2000, 11 µM cisplatin | 4394 |
| A549rCDDP2000, untreated | A549rCDDP2000, 11 µM cisplatin | 27 |
| A549rCDDP2000, untreated | A549rCDDP2000, 34 µM cisplatin | 708 |
| A549, untreated | A549, 11 µM cisplatin | 1191 |
| A549, 11 µM cisplatin | A549rCDDP2000, 34 µM cisplatin | 3670 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarin, N.; Engel, F.; Rothweiler, F.; Cinatl, J.; Michaelis, M.; Frötschl, R.; Fröhlich, H.; Kalayda, G.V. Key Players of Cisplatin Resistance: Towards a Systems Pharmacology Approach. Int. J. Mol. Sci. 2018, 19, 767. https://doi.org/10.3390/ijms19030767
Sarin N, Engel F, Rothweiler F, Cinatl J, Michaelis M, Frötschl R, Fröhlich H, Kalayda GV. Key Players of Cisplatin Resistance: Towards a Systems Pharmacology Approach. International Journal of Molecular Sciences. 2018; 19(3):767. https://doi.org/10.3390/ijms19030767
Chicago/Turabian StyleSarin, Navin, Florian Engel, Florian Rothweiler, Jindrich Cinatl, Martin Michaelis, Roland Frötschl, Holger Fröhlich, and Ganna V. Kalayda. 2018. "Key Players of Cisplatin Resistance: Towards a Systems Pharmacology Approach" International Journal of Molecular Sciences 19, no. 3: 767. https://doi.org/10.3390/ijms19030767