The Role of Macrophages in Neuroinflammatory and Neurodegenerative Pathways of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis: Pathogenetic Cellular Effectors and Potential Therapeutic Targets

 , , ,
, , ,
Abstract
:1. Introduction
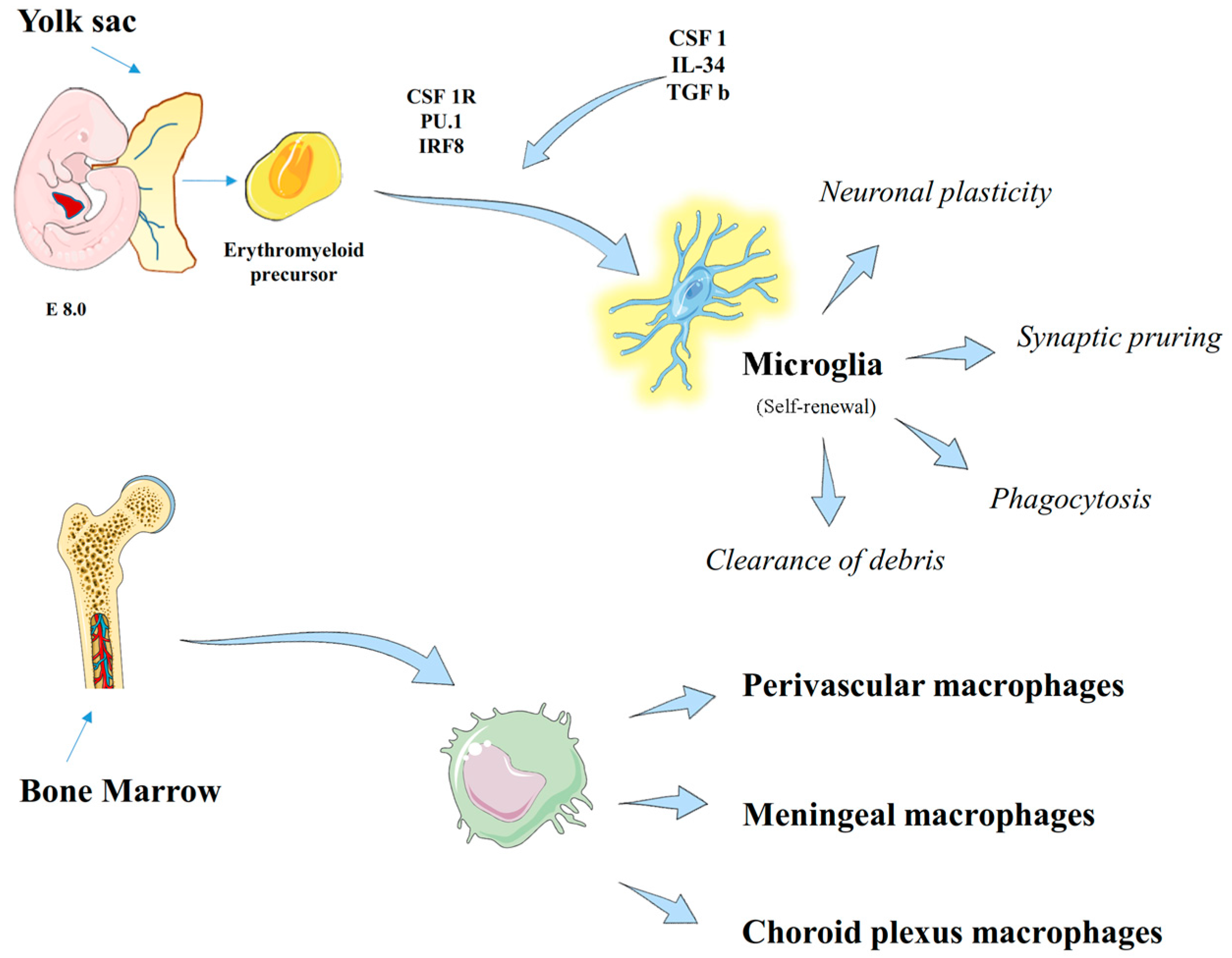
2. Macrophage Populations in the Central Nervous System
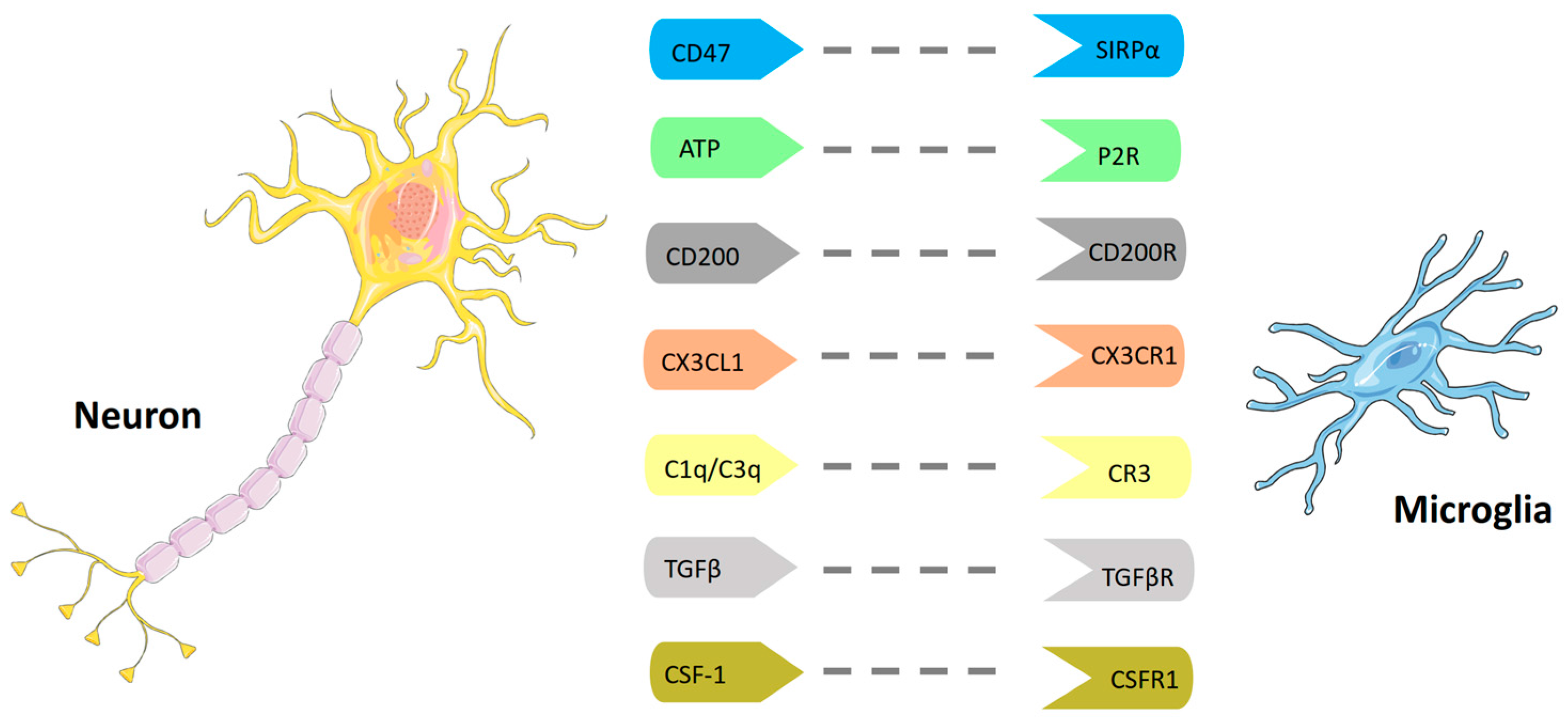
2.1. Microglia Physiology
2.2. Microglia in CNS Pathology
2.3. Non-Parenchymal CNS Macrophages
3. Central Nervous System Macrophages in the Pathogenesis of Neuroinflammatory/Neurodegenerative Diseases
3.1. Alzheimer’s Disease
3.2. Amyotrophic Lateral Sclerosis
3.3. Multiple Sclerosis
3.4. Myeloid-Targeted Therapy
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CNS | Central nervous system |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| MS | Multiple sclerosis |
| HSCs | hematopoietic cells |
| CSF1R | CSF1 receptor |
| IL-34 | Interleukin-34 |
| CNS-TGF-β1−/− | mice deficient in TGF-β1 in the CNS |
| TMEM119 | transmembrane protein 119 |
| Siglec-H | sialic acid-binding immunoglobulin-like lectin H |
| P2RY12 | P2Y purinoceptor 12 |
| SALL1 | Sal-like protein 1 |
| TGFβ1 | transforming growth factor-β1 |
| TGFβR1 | TGFβ receptor 1 |
| BDNF | Brain-derived neurotrophic factor |
| GDNF | Glial cell-derived neurotrophic factor |
| NGF | Nerve growth factor |
| MCHII | major histocompatibility class II |
| MOG | Myelin oligodendrocyte glycoprotein |
| EAE | Experimental allergic encephalomyelitis |
| DAMPs | damage-associated molecular patterns |
| CCR2 | C-C chemokine receptor 2 |
| Fc | fragment crystallizable |
| SIRPα | signal regulatory protein α |
| RAGE | Receptor for advanced glycation end-products |
| IL | Interleukin |
| iNOS | inducible nitric oxide synthase |
| Th1 | T helper 1 (Th1) |
| IFNγ | interferon-γ |
| LPS | lipopolysaccharide |
| TLR | Toll-like receptor |
| Psen1 | Presenilin-1 |
| Psen2 | Presenilin-2 |
| ApoE | Apolipoprotein E |
| APP | amyloid precursor protein |
| NSAIDs | non-steroidal anti-inflammatory drugs |
| DAP12 | DNAX-activating protein 12 |
| SOD1 | superoxide dismutase 1 |
| FUS | Fused in Sarcoma |
| TDP-43 | TAR DNA-binding protein 43 |
| mSOD1 | mutant SOD1 |
| PU.1−/− | PU.1 knockout |
| BBG | Brilliant Blue G |
| DC | dendritic cells |
| ROS | reactive oxygen species |
| TNF | tumor necrosis factor |
| TGF | transforming growth factor |
| TAK1 | transforming growth factor (TGF)-β-activated kinase 1 |
| Sh | short hairpin |
| AMPK | AMP-activated protein kinase |
| CaMKKβ | calmodulin-dependent protein kinase kinase β |
| GSK3β | glycogen synthase kinase-3 β |
| PI3K | The phosphatidylinositol 3-kinase |
| CBD | cannabidiol |
| PMMA-NPs | poly(methyl methacrylate) nanoparticles |
| rAAV | recombinant vectors based on adeno-associated virus |
References
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS immune privilege: Hiding in plain sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J.; Sisodia, S.S.; Ransohoff, R.M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 2011, 14, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Huang, A.; Wen, S.; Liao, B.; Appel, S.H. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 2011, 134, 1293–1314. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Mittelbronn, M.; Dietz, K.; Schluesener, H.J.; Meyermann, R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001, 101, 249–255. [Google Scholar] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Perdiguero, E.G.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A lineage of myeloid cells independent of myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhang, P.; Hirai, H.; Elf, S.; Yan, X.; Chen, Z.; Koschmieder, S.; Okuno, Y.; Dayaram, T.; Growney, J.D.; et al. PU.1 is a major downstream target of AML1 (RUNX1) in adult mouse hematopoiesis. Nat. Genet. 2008, 40, 51–60. [Google Scholar] [CrossRef] [PubMed]
- McKercher, S.R.; Torbett, B.E.; Anderson, K.L.; Henkel, G.W.; Vestal, D.J.; Baribault, H.; Klemsz, M.; Feeney, A.J.; Wu, G.E.; Paige, C.J.; et al. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996, 15, 5647–5658. [Google Scholar] [PubMed]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [PubMed]
- Greter, M.; Lelios, I.; Pelczar, P.; Hoeffel, G.; Price, J.; Leboeuf, M.; Kündig, T.M.; Frei, K.; Ginhoux, F.; Merad, M.; Becher, B. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 2012, 37, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738-46. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Kobayashi, M.; Kunisawa, T.; Imai, K.; Sayo, A.; Malissen, B.; Crocker, P.R.; Sato, K.; Kiyama, H. Siglec-H is a microglia-specific marker that discriminates microglia from CNS-associated macrophages and CNS-infiltrating monocytes. Glia 2017, 65, 1927–1943. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, E.M.; Cochrane, F.; Barclay, A.N.; van den Berg, T.K. Signal regulatory proteins in the immune system. J. Immunol. 2005, 175, 7781–7787. [Google Scholar] [CrossRef] [PubMed]
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 2017, 18, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Shemer, A.; Erny, D.; Jung, S.; Prinz, M. Microglia plasticity during health and disease: An immunological perspective. Trends Immunol. 2015, 36, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernández-Klett, F.; Lin, G.; Sagar; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.; Han, S.J.; Kaur, G.; Crane, C.; Parsa, A.T. The role of microglia in central nervous system immunity and glioma immunology. J. Clin. Neurosci. 2010, 17, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Semple, B.D.; Kossmann, T.; Morganti-Kossmann, M.C. Role of chemokines in CNS health and pathology: A focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab. 2010, 30, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Barclay, A.N.; Wright, G.J.; Brooke, G.; Brown, M.H. CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 2002, 23, 285–290. [Google Scholar] [CrossRef]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W.G.M. Neuronal “On” and “Off” signals control microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Banisadr, G.; Quéraud-Lesaux, F.; Boutterin, M.C.; Pélaprat, D.; Zalc, B.; Rostène, W.; Haour, F.; Parsadaniantz, S.M. Distribution, cellular localization and functional role of CCR2 chemokine receptors in adult rat brain. J. Neurochem. 2002, 81, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Boddeke, E.W.; Meigel, I.; Frentzel, S.; Gourmala, N.G.; Harrison, J.K.; Buttini, M.; Spleiss, O.; Gebicke-Härter, P. Cultured rat microglia express functional β-chemokine receptors. J. Neuroimmunol. 1999, 98, 176–184. [Google Scholar] [CrossRef]
- Sivakumar, V.; Foulds, W.S.; Luu, C.D.; Ling, E.-A.; Kaur, C. Retinal ganglion cell death is induced by microglia derived pro-inflammatory cytokines in the hypoxic neonatal retina. J. Pathol. 2011, 224, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shi, X.Q.; Echeverry, S.; Mogil, J.S.; De Koninck, Y.; Rivest, S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J. Neurosci. 2007, 27, 12396–12406. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Y.; Lu, J.; Ling, E.A.; Kaur, C. Monocyte chemoattractant protein-1 (MCP-1) produced via NF-kappaB signaling pathway mediates migration of amoeboid microglia in the periventricular white matter in hypoxic neonatal rats. Glia 2009, 57, 604–621. [Google Scholar] [CrossRef] [PubMed]
- Dimitrijevic, O.B.; Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 2007, 38, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kellner, C.P.; Hahn, D.K.; Desantis, B.M.; Musabbir, M.; Starke, R.M.; Rynkowski, M.; Komotar, R.J.; Otten, M.L.; Sciacca, R.; et al. Monocyte chemoattractant protein-1 predicts outcome and vasospasm following aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2008, 109, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M. ATP: A ubiquitous gliotransmitter integrating neuron–glial networks. Semin. Cell Dev. Biol. 2011, 22, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Domercq, M.; Vázquez-Villoldo, N.; Matute, C. Neurotransmitter signaling in the pathophysiology of microglia. Front. Cell. Neurosci. 2013, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Ohsawa, K.; Inoue, K.; Kohsaka, S. Purinergic receptors in microglia: Functional modal shifts of microglia mediated by P2 and P1 receptors. Glia 2013, 61, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, W.; Li, Q.; Tang, M. Extracellular nucleotides and adenosine regulate microglial motility and their role in cerebral ischemia. Acta Pharm. Sin. B 2013, 3, 205–212. [Google Scholar] [CrossRef]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [PubMed]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.; Koenigsknecht-Talboo, J.; Grommes, C.; Lee, C.Y.D.; Landreth, G. Fibrillar β-amyloid-stimulated intracellular signaling cascades require VAV for induction of respiratory burst and phagocytosis in monocytes and microglia. J. Biol. Chem. 2006, 281, 20842–20850. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Shapiro, S.; Goldman, D.L.; Casadevall, A.; Scharff, M.; Lee, S.C. Fcγ receptor I- and III-mediated macrophage inflammatory protein 1α induction in primary human and murine microglia. Infect. Immun. 2002, 70, 5177–5184. [Google Scholar] [CrossRef] [PubMed]
- Gresham, H.D.; Dale, B.M.; Potter, J.W.; Chang, P.W.; Vines, C.M.; Lowell, C.A.; Lagenaur, C.F.; Willman, C.L. Negative regulation of phagocytosis in murine macrophages by the SRC kinase family member, FGR. J. Exp. Med. 2000, 191, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Oldenborg, P.A.; Gresham, H.D.; Lindberg, F.P. CD47-signal regulatory protein α (SIRPα) regulates Fcγ and complement receptor-mediated phagocytosis. J. Exp. Med. 2001, 193, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Lagenaur, C.F.; Narayanan, V. Integrin-associated protein is a ligand for the P84 neural adhesion molecule. J. Biol. Chem. 1999, 274, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Dangott, L.J.; Pfenninger, K.H. The heterogeneous growth cone glycoprotein gp93 is identical to the signal regulatory protein SIRPα/SHPS-1/BIT. J. Neurochem. 2003, 86, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Han, M.H.; Lundgren, D.H.; Jaiswal, S.; Chao, M.; Graham, K.L.; Garris, C.S.; Axtell, R.C.; Ho, P.P.; Lock, C.B.; Woodard, J.I.; et al. Janus-like opposing roles of CD47 in autoimmune brain inflammation in humans and mice. J. Exp. Med. 2012, 209, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Bucciarelli, L.G.; Wendt, T.; Sakaguchi, T.; Lalla, E.; Qu, W.; Lu, Y.; Lee, L.; Stern, D.M.; Naka, Y.; et al. Blockade of receptor for advanced glycation endproducts: A new target for therapeutic intervention in diabetic complications and inflammatory disorders. Arch. Biochem. Biophys. 2003, 419, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J. Transl. Med. 2009, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, S.; Barakat, W.; Stoyanov, S.; Murikinati, S.; Yang, H.; Tracey, K.J.; Bendszus, M.; Rossetti, G.; Nawroth, P.P.; Bierhaus, A.; et al. The HMGB1 receptor RAGE mediates ischemic brain damage. J. Neurosci. 2008, 28, 12023–12031. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Zaccone, P.; Di Marco, R.; Lunetta, M.; Magro, G.; Grasso, S.; Meroni, P.; Garotta, G. Prevention of spontaneous autoimmune diabetes in diabetes-prone BB rats by prophylactic treatment with antirat interferon-γ antibody. Endocrinology 1997, 138, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.S.; Hemmerle, T.; Pretto, F.; Kipar, A.; Neri, D. Antibody-based targeted delivery of interleukin-4 synergizes with dexamethasone for the reduction of inflammation in arthritis. Rheumatology 2018. [Google Scholar] [CrossRef] [PubMed]
- De Waal Malefyt, R.; Abrams, J.; Bennett, B.; Figdor, C.G.; de Vries, J.E. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 1991, 174, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Type 2 cytokines: Mechanisms and therapeutic strategies. Nat. Rev. Immunol. 2015, 15, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Galea, I.; Palin, K.; Newman, T.A.; Van Rooijen, N.; Perry, V.H.; Boche, D. Mannose receptor expression specifically reveals perivascular macrophages in normal, injured, and diseased mouse brain. Glia 2005, 49, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Fabriek, B.O.; Van Haastert, E.S.; Galea, I.; Polfliet, M.M.J.; Döpp, E.D.; Van Den Heuvel, M.M.; Van Den Berg, T.K.; De Groot, C.J.A.; Van Der Valk, P.; Dijkstra, C.D. CD163-positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia 2005, 51, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.X.; Li, J.; Ventura, E.; Rostami, A. Parenchymal microglia of naïve adult C57BL/6J mice express high levels of B7.1, B7.2, and MHC class II. Exp. Mol. Pathol. 2002, 73, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Polfliet, M.M.; Zwijnenburg, P.J.; van Furth, A.M.; van der Poll, T.; Döpp, E.A.; Renardel de Lavalette, C.; van Kesteren-Hendrikx, E.M.; van Rooijen, N.; Dijkstra, C.D.; van den Berg, T.K. Meningeal and perivascular macrophages of the central nervous system play a protective role during bacterial meningitis. J. Immunol. 2001, 167, 4644–4650. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Muramatsu, M.; Azuma, E.; Ikutani, M.; Nagai, Y.; Sagara, H.; Koo, B.-N.; Kita, S.; O’Donnell, E.; Osawa, T.; et al. A subset of cerebrovascular pericytes originates from mature macrophages in the very early phase of vascular development in CNS. Sci. Rep. 2017, 7, 3855. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.-C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of β-amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, W.; Wen, W.; Sun, J.; Su, B.; Liu, B.; Ma, D.; Lv, D.; Wen, Y.; Qu, T.; et al. Secretase Gene Mutations in Familial Acne Inversa. Science 2010, 330, 1065. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the β-amyloid precursor protein in familial Alzheimer’s disease increases β-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Holtzman, D.M. Apolipoprotein E levels and Alzheimer risk. Ann. Neurol. 2015, 77, 204–205. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Rogers, J. Anti-inflammatory agents as a therapeutic approach to Alzheimer’s disease. Neurology 1992, 42, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.M.; Vorbrodt, A.W.; Wegiel, J.; Morys, J.; Lossinsky, A.S. Ultrastructure of the cells forming amyloid fibers in Alzheimer disease and scrapie. Am. J. Med. Genet. Suppl. 1990, 7, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chen, W.-D.; Wang, Y.-D. β-Amyloid: The key peptide in the pathogenesis of Alzheimer’s disease. Front. Pharmacol. 2015, 6, 221. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.H.; Bigbee, M.J.; Boynton, G.E.; Wakeham, C.M.; Rosenheim, H.M.; Staral, C.J.; Morrissey, J.L.; Hund, A.K. Non-Steroidal Anti-Inflammatory Drugs in Alzheimer’s Disease and Parkinson’s Disease: Reconsidering the Role of Neuroinflammation. Pharmaceuticals 2010, 3, 1812–1841. [Google Scholar] [CrossRef] [PubMed]
- Van Eldik, L.J.; Carrillo, M.C.; Cole, P.E.; Feuerbach, D.; Greenberg, B.D.; Hendrix, J.A.; Kennedy, M.; Kozauer, N.; Margolin, R.A.; Molinuevo, J.L.; et al. The roles of inflammation and immune mechanisms in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2016, 2, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Yamasaki, T.R.; LaFerla, F.M. Microglia as a potential bridge between the amyloid β-peptide and tau. Ann. N. Y. Acad. Sci. 2004, 1035, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Solito, E.; Sastre, M. Microglia function in Alzheimer’s disease. Front. Pharmacol. 2012, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.-P.; Rivest, S. Bone Marrow-Derived Microglia Play a Critical Role in Restricting Senile Plaque Formation in Alzheimer’s Disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Giulian, D. Microglia and the immune pathology of Alzheimer disease. Am. J. Hum. Genet. 1999, 65, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht, J.; Landreth, G. Microglial Phagocytosis of Fibrillar β-Amyloid through a β-1 Integrin-Dependent Mechanism. J. Neurosci. 2004, 24, 9838–9846. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.-S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; Von Korff, A.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid β. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Jia, L.; Liu, C.-C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.-F.; Fryer, J.D.; Wang, X.; Zhang, Y.; et al. TREM2 Promotes Microglial Survival by Activating Wnt/β-Catenin Pathway. J. Neurosci. 2017, 37, 1772–1784. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, G.; Yamanishi, Y.; Suárez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243ra86. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.E.; Luster, A.D. Mechanisms of microglia accumulation in Alzheimer’s disease: Therapeutic implications. Trends Pharmacol. Sci. 2008, 29, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Thériault, P.; ElAli, A.; Rivest, S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimer’s Res. Ther. 2015, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Varvel, N.H.; Konerth, M.E.; Xu, G.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T. CX3CR1 deficiency alters microglial activation and reduces β-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 2010, 177, 2549–2562. [Google Scholar] [CrossRef] [PubMed]
- Parvathenani, L.K.; Tertyshnikova, S.; Greco, C.R.; Roberts, S.B.; Robertson, B.; Posmantur, R. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J. Biol. Chem. 2003, 278, 13309–13317. [Google Scholar] [CrossRef] [PubMed]
- McLarnon, J.G.; Ryu, J.K.; Walker, D.G.; Choi, H.B. Upregulated Expression of Purinergic P2X7 Receptor in Alzheimer Disease and Amyloid-β Peptide-Treated Microglia and in Peptide-Injected Rat Hippocampus. J. Neuropathol. Exp. Neurol. 2006, 65, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wang, P.; Zhang, J.; Chen, W.; Gu, L. Silencing of the P2X7 receptor enhances amyloid-β phagocytosis by microglia. Biochem. Biophys. Res. Commun. 2013, 434, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Ajit, D.; Woods, L.T.; Camden, J.M.; Thebeau, C.N.; El-Sayed, F.G.; Greeson, G.W.; Erb, L.; Petris, M.J.; Miller, D.C.; Sun, G.Y.; et al. Loss of P2Y2 nucleotide receptors enhances early pathology in the TgCRND8 mouse model of Alzheimer’s disease. Mol. Neurobiol. 2014, 49, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; McLaurin, J. Selective targeting of perivascular macrophages for clearance of β-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Puentes, F.; Malaspina, A.; van Noort, J.M.; Amor, S. Non-neuronal Cells in ALS: Role of Glial, Immune cells and Blood-CNS Barriers. Brain Pathol. 2016, 26, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.-A.; Manning, J.; Rossi, F.; Krieger, C. The neuroinflammatory response in ALS: The roles of microglia and T cells. Neurol. Res. Int. 2012, 2012, 803701. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Zhao, W.; Beers, D.R.; Henkel, J.S.; Appel, S.H. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 2012, 237, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Siddiqui, S.; Gabriely, G.; Lanser, A.J.; Dake, B.; Murugaiyan, G.; Doykan, C.E.; Wu, P.M.; Gali, R.R.; Iyer, L.K.; et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J. Clin. Investig. 2012, 122, 3063–3087. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosi, N.; Finocchi, P.; Apolloni, S.; Cozzolino, M.; Ferri, A.; Padovano, V.; Pietrini, G.; Carri, M.T.; Volonte, C. The proinflammatory action of microglial P2 receptors is enhanced in SOD1 models for amyotrophic lateral sclerosis. J. Immunol. 2009, 183, 4648–4656. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, S.; Amadio, S.; Parisi, C.; Matteucci, A.; Potenza, R.L.; Armida, M.; Popoli, P.; D’Ambrosi, N.; Volonte, C. Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. Dis. Model. Mech. 2014, 7, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Høglund, R.A.; Maghazachi, A.A. Multiple sclerosis and the role of immune cells. World J. Exp. Med. 2014, 4, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shemer, A.; Jung, S. Differential roles of resident microglia and infiltrating monocytes in murine CNS autoimmunity. Semin. Immunopathol. 2015, 37, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of “homeostatic” microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Kocur, M.; Schneider, R.; Pulm, A.-K.; Bauer, J.; Kropp, S.; Gliem, M.; Ingwersen, J.; Goebels, N.; Alferink, J.; Prozorovski, T.; et al. IFNβ secreted by microglia mediates clearance of myelin debris in CNS autoimmunity. Acta Neuropathol. Commun. 2015, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Teige, I.; Treschow, A.; Teige, A.; Mattsson, R.; Navikas, V.; Leanderson, T.; Holmdahl, R.; Issazadeh-Navikas, S. IFN-β gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J. Immunol. 2003, 170, 4776–4784. [Google Scholar] [CrossRef] [PubMed]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.-M.; Sánchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.-È.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.J.; Polak, P.E.; Simonini, V.; Lin, S.X.; Richardson, J.C.; Bongarzone, E.R.; Feinstein, D.L. P2x7 deficiency suppresses development of experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2008, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Matute, C.; Torre, I.; Perez-Cerda, F.; Perez-Samartin, A.; Alberdi, E.; Etxebarria, E.; Arranz, A.M.; Ravid, R.; Rodriguez-Antiguedad, A.; Sanchez-Gomez, M.; et al. P2X7 receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 2007, 27, 9525–9533. [Google Scholar] [CrossRef] [PubMed]
- Mélik-Parsadaniantz, S.; Rostène, W. Chemokines and neuromodulation. J. Neuroimmunol. 2008, 198, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Hulkower, K.; Brosnan, C.F.; Aquino, D.A.; Cammer, W.; Kulshrestha, S.; Guida, M.P.; Rapoport, D.A.; Berman, J.W. Expression of CSF-1, c-fms, and MCP-1 in the central nervous system of rats with experimental allergic encephalomyelitis. J. Immunol. 1993, 150, 2525–2533. [Google Scholar] [PubMed]
- Dogan, R.-N.E.; Elhofy, A.; Karpus, W.J. Production of CCL2 by central nervous system cells regulates development of murine experimental autoimmune encephalomyelitis through the recruitment of TNF- and iNOS-expressing macrophages and myeloid dendritic cells. J. Immunol. 2008, 180, 7376–7384. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Niemi, E.C.; Wang, S.H.; Lee, C.C.; Bingham, D.; Zhang, J.; Cozen, M.L.; Charo, I.; Huang, E.J.; Liu, J.; et al. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J. Neurotrauma 2014, 31, 1677–1688. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Bannerman, P.; Ma, J.; Guo, F.; Miers, L.; Soulika, A.M.; Pleasure, D. Conditional Ablation of Astroglial CCL2 Suppresses CNS Accumulation of M1 Macrophages and Preserves Axons in Mice with MOG Peptide EAE. J. Neurosci. 2014, 34, 8175–8185. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Wieghofer, P.; Müller, P.F.; Wolf, Y.; Varol, D.; Yona, S.; Brendecke, S.M.; Kierdorf, K.; Staszewski, O.; Datta, M.; et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 2013, 16, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-B.; Sig Choi, J.; Yu, Y.-M.; Nam, K.; Piao, C.-S.; Kim, S.-W.; Lee, M.-H.; Han, P.-L.; Park, J.-S.; Lee, J.-K. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 2006, 26, 6413–6421. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Kitamura, Y.; Tsuchiya, D.; Kawasaki, T.; Taniguchi, T.; Shimohama, S. High mobility group box protein-1 inhibits microglial Aβ clearance and enhances Aβ neurotoxicity. J. Neurosci. Res. 2004, 78, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.-S. HMGB1 acts on microglia MAC1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Mollica, L.; De Marchis, F.; Spitaleri, A.; Dallacosta, C.; Pennacchini, D.; Zamai, M.; Agresti, A.; Trisciuoglio, L.; Musco, G.; Bianchi, M.E. Glycyrrhizin Binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem. Biol. 2007, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.-Y.; Tang, C.-H.; Chen, Y.-H.; Wei, I.-H. Berberine suppresses neuroinflammatory responses through AMP-activated protein kinase activation in BV-2 microglia. J. Cell. Biochem. 2010, 110, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, J.; Shi, J.-S. Anti-inflammatory activities of resveratrol in the brain: Role of resveratrol in microglial activation. Eur. J. Pharmacol. 2010, 636, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Haghani, M.; Shabani, M.; Tondar, M. The therapeutic potential of berberine against the altered intrinsic properties of the CA1 neurons induced by Aβ neurotoxicity. Eur. J. Pharmacol. 2015, 758, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Meares, G.P.; Qin, H.; Liu, Y.; Holdbrooks, A.T.; Benveniste, E.N. AMP-activated protein kinase restricts IFN-γ signaling. J. Immunol. 2013, 190, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xu, Y.; Wang, Y.; Wang, Y.; He, L.; Jiang, Z.; Huang, Z.; Liao, H.; Li, J.; Saavedra, J.M.; et al. Telmisartan prevention of LPS-induced microglia activation involves M2 microglia polarization via CaMKKβ-dependent AMPK activation. Brain. Behav. Immun. 2015, 50, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Erbe, D.V.; Gartrell, K.; Zhang, Y.-L.; Suri, V.; Kirincich, S.J.; Will, S.; Perreault, M.; Wang, S.; Tobin, J.F. Molecular activation of PPARγ by angiotensin II type 1-receptor antagonists. Vascul. Pharmacol. 2006, 45, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.; Wang, J.; Benicky, J.; Sánchez-Lemus, E.; Saavedra, J.M. Telmisartan directly ameliorates the neuronal inflammatory response to IL-1β partly through the JNK/c-Jun and NADPH oxidase pathways. J. Neuroinflamm. 2012, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pang, T.; Hafko, R.; Benicky, J.; Sanchez-Lemus, E.; Saavedra, J.M. Telmisartan ameliorates glutamate-induced neurotoxicity: Roles of AT(1) receptor blockade and PPARγ activation. Neuropharmacology 2014, 79, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Masciopinto, F.; Di Pietro, N.; Corona, C.; Bomba, M.; Pipino, C.; Curcio, M.; di Castelnuovo, A.; Ciavardelli, D.; Silvestri, E.; Canzoniero, L.M.T.; et al. Effects of long-term treatment with pioglitazone on cognition and glucose metabolism of PS1-KI, 3xTg-AD, and wild-type mice. Cell Death Dis. 2012, 3, e448. [Google Scholar] [CrossRef] [PubMed]
- Schütz, B.; Reimann, J.; Dumitrescu-Ozimek, L.; Kappes-Horn, K.; Landreth, G.E.; Schürmann, B.; Zimmer, A.; Heneka, M.T. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J. Neurosci. 2005, 25, 7805–7812. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Dengler, R.; Heneka, M.T.; Meyer, T.; Zierz, S.; Kassubek, J.; Fischer, W.; Steiner, F.; Lindauer, E.; Otto, M.; et al. A randomized, double blind, placebo-controlled trial of pioglitazone in combination with riluzole in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e37885. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [PubMed]
- Yuskaitis, C.J.; Jope, R.S. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 2009, 21, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.-H.; Kim, Y.; Kim, H.Y.; Hwang, S.; Lee, C.H.; Kim, S.H. Inhibition of glycogen synthase kinase-3 suppresses the onset of symptoms and disease progression of G93A-SOD1 mouse model of ALS. Exp. Neurol. 2007, 205, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.-W.; Jeon, G.S.; Kim, M.-J.; Shon, J.-H.; Kim, J.-E.; Shin, J.-Y.; Kim, S.-M.; Kim, S.H.; Ye, I.-H.; Lee, K.-W.; et al. Neuroprotective effects of JGK-263 in transgenic SOD1-G93A mice of amyotrophic lateral sclerosis. J. Neurol. Sci. 2014, 340, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Stella, N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Lourbopoulos, A.; Grigoriadis, N.; Lagoudaki, R.; Touloumi, O.; Polyzoidou, E.; Mavromatis, I.; Tascos, N.; Breuer, A.; Ovadia, H.; Karussis, D.; et al. Administration of 2-arachidonoylglycerol ameliorates both acute and chronic experimental autoimmune encephalomyelitis. Brain Res. 2011, 1390, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Martín-Moreno, A.M.; Reigada, D.; Ramírez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; de Ceballos, M.L. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: Relevance to Alzheimer’s disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Monji, A.; Hashioka, S.; Kanba, S. Risperidone significantly inhibits interferon-γ-induced microglial activation in vitro. Schizophr. Res. 2007, 92, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, H.; Kato, T.A.; Mizoguchi, Y.; Monji, A.; Seki, Y.; Ohkuri, T.; Gotoh, L.; Yonaha, M.; Ueda, T.; Hashioka, S.; et al. Inhibitory effects of SSRIs on IFN-γ induced microglial activation through the regulation of intracellular calcium. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Bian, Q.; Kato, T.; Monji, A.; Hashioka, S.; Mizoguchi, Y.; Horikawa, H.; Kanba, S. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-γ. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Ferrari, R.; De Paola, M.; Rossi, F.; Mariani, A.; Caron, I.; Sammali, E.; Peviani, M.; Dell’Oro, V.; Colombo, C.; et al. Polymeric nanoparticle system to target activated microglia/macrophages in spinal cord injury. J. Control. Release 2014, 174, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Cucchiarini, M.; Ren, X.L.; Perides, G.; Terwilliger, E.F. Selective gene expression in brain microglia mediated via adeno-associated virus type 2 and type 5 vectors. Gene Ther. 2003, 10, 657–667. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Treatment | Target | Disease | Effect | References |
|---|---|---|---|---|
| Inflachromene | HMGB1–HMGB2 | Animal Model of AD | Blocks post-translational modifications and releases reduce microglial activation | [126,127] |
| Berberine Resveratrol | AMPK AMPK | LPS- and IFN γ BV-2 microglia cells Animal model of AD Animal model of AD | Reduces neuroinflammation Ameliorates neurotoxicity induced by Aβ. Improves the cognitive impairment | [129,130,131] |
| Telmisartan | AMPK PPARγ | LPS-challenged microglia cell | Promotes M2 polarization and reduces M1 polarization Reduction of neuronal injury and microglia activation | [133] |
| Pioglitazone | PPARγ | Animal model of ALS Animal models of AD | Reduces neuron damage and increases survival Reduces neuroinflammation, Improves disease severity | [137,138,150] |
| GSK3β inhibitors | GSK3β | LPS-challenged BV-2 microglia cell Animal model of ALS | Reduces IL-6 and NO Delays onset of symptoms and prolongs the lifespan | [141,142,143] |
| 2-arachydonyl-glycerol Cannabidiol (CBD) | Endocannabinoid receptor | Experimental Allergic Encephalomyelitis Animal model of AD | Improves disease course Prevents the cognitive impairment | [145,146] |
| Risperidone, Perospirone and Quetiapine | D2 receptor | IFN γ activated microglia cells | Suppresses the release of pro-inflammatory cytokines | [147,148,149] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mammana, S.; Fagone, P.; Cavalli, E.; Basile, M.S.; Petralia, M.C.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The Role of Macrophages in Neuroinflammatory and Neurodegenerative Pathways of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis: Pathogenetic Cellular Effectors and Potential Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 831. https://doi.org/10.3390/ijms19030831
Mammana S, Fagone P, Cavalli E, Basile MS, Petralia MC, Nicoletti F, Bramanti P, Mazzon E. The Role of Macrophages in Neuroinflammatory and Neurodegenerative Pathways of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis: Pathogenetic Cellular Effectors and Potential Therapeutic Targets. International Journal of Molecular Sciences. 2018; 19(3):831. https://doi.org/10.3390/ijms19030831
Chicago/Turabian StyleMammana, Santa, Paolo Fagone, Eugenio Cavalli, Maria Sofia Basile, Maria Cristina Petralia, Ferdinando Nicoletti, Placido Bramanti, and Emanuela Mazzon. 2018. "The Role of Macrophages in Neuroinflammatory and Neurodegenerative Pathways of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis: Pathogenetic Cellular Effectors and Potential Therapeutic Targets" International Journal of Molecular Sciences 19, no. 3: 831. https://doi.org/10.3390/ijms19030831