
Abstract
Trypanosomes are masters of adaptation to different host environments during their complex life cycle. Large-scale proteomic approaches provide information on changes at the cellular level, and in a systematic way. However, detailed work on single components is necessary to understand the adaptation mechanisms on a molecular level. Here, we have performed a detailed characterization of a bloodstream form (BSF) stage-specific putative flagellar host adaptation factor Tb927.11.2400, identified previously in a SILAC-based comparative proteome study. Tb927.11.2400 shares 38% amino acid identity with TbFlabarin (Tb927.11.2410), a procyclic form (PCF) stage-specific flagellar BAR domain protein. We named Tb927.11.2400 TbFlabarin-like (TbFlabarinL), and demonstrate that it originates from a gene duplication event, which occurred in the African trypanosomes. TbFlabarinL is not essential for the growth of the parasites under cell culture conditions and it is dispensable for developmental differentiation from BSF to the PCF in vitro. We generated TbFlabarinL-specific antibodies, and showed that it localizes in the flagellum. Co-immunoprecipitation experiments together with a biochemical cell fractionation suggest a dual association of TbFlabarinL with the flagellar membrane and the components of the paraflagellar rod.
Similar content being viewed by others
Introduction
The Kinetoplastida are a class of unicellular protists that share a common feature (the kinetoplast), comprising the mitochondrial DNA of the cell1. The class Kinetoplastida includes the order of exclusively parasitic Trypanosomatida, which are either monoxenous (restricted to one host individual), or dixenous (undergoing a complex life cycle between a host and a vector). Some species of this order are the causative agents of various infectious diseases distributed in many parts of the world, with a devastating impact on human health and the economies of impoverished countries. For example, Leishmania donovani causes visceral leishmaniasis (Kala-Azar) in South and Central America, South Europe, Africa and West Asia; Trypanosoma cruzi causes Chagas disease in South America; Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense are responsible for sleeping sickness in humans (human African trypanosomiasis, HAT), and Trypanosoma brucei brucei causes nagana in livestock in sub-Saharan Africa2. Both Leishmania and the two Trypanosoma species are transmitted to mammals by bloodsucking insects.
T. brucei has developed a complicated life cycle with different developmental stages in order to proliferate in mammalian hosts and to use the tsetse fly for dissemination. The T. brucei parasites first proliferate in the blood and adipose tissue of infected mammals as a long slender (LS) bloodstream form (BSF)3. The surface of the BSF parasites is covered by a densely packed variant surface glycoprotein (VSG) coat, which is central to the antigenic variation mechanism contributing to host immune system evasion4. Upon accumulation of a quorum sensing signal called stumpy induction factor (SIF)5, LS parasites differentiate into a cell cycle-arrested short stumpy (SS) form, which are pre-adapted for further differentiation in the tsetse fly. In the fly midgut, the SS form differentiates into a dividing procyclic form (PCF), the surface of which is covered with procyclin6. While PCFs migrate from the midgut they further differentiate and colonize the salivary glands as epimastigotes. Epimastigote trypanosomes are able to attach to the host microvilli of the epithelial cells lining the salivary glands lumen by the flagellar membrane flagellipodia7. While still attached to the salivary gland surface, trypanosomes acquire the VSG coat and mature into metacyclics. These cells are preadapted for transfer to and life in the mammalian host when the fly takes a next blood meal and completes the life cycle of the parasite8.
The mammalian host and the tsetse fly vector represent two completely different environments in terms of host immune response challenges, energy resources and temperature. The parasite has developed a sophisticated adaptation strategy and adjusts its morphology, motility, metabolism, gene expression and organelle activity to survive and proliferate in these different host environments. More recently, it was shown that even within the mammalian host, the parasites adapt to the different tissues, as parasites in the blood and adipose tissue are functionally different3. Understanding the host adaptation mechanisms of T. brucei during its life cycle has been a challenging task for the field. Several genome-wide transcriptome analyses have been performed to elucidate how trypanosomes adapt to different host environments. In addition to the comparison of transcript abundance in PCF and BSF9, the transcriptome of differentiating parasites has been analyzed10,11.
These studies provided many insights into the adaptation machinery of trypanosomes but there are certain limitations to transcriptome-based approaches. Due to the fact that the regulation of gene expression in trypanosomes occurs almost exclusively post-transcriptionally–at the level of mRNA stability, translational efficiency, and protein stability–the levels of mRNA do not always reflect the actual protein abundance in the cell12. For example, transcriptome-wide quantification of mRNA stability revealed that highly abundant transcripts in BSF have longer half lives compared to the same transcripts in PCF13. Furthermore, translational efficiency in PCF and BSF varies greatly between these two life cycle stages as shown by ribosome profiling14,15.
Hence, proteome-based studies are required to completely understand how the parasite changes during developmental differentiation. Recently, stable isotope labeling (SILAC) was used to quantitatively compare the proteomes of BSF and PCF, which elucidated many new components of the machinery for adaptation to the insect and mammalian hosts16,17,18. A previous study from our laboratories elucidated many new components of the host adaptation machinery16. A total of 4364 protein groups were analyzed and many new putative proteins of unknown function were detected. In all, 625 protein groups were enriched in the PCF and 253 protein groups were enriched in the BSF16. Furthermore, we also used label-free mass spectrometry techniques to quantify changes of the trypanosome proteome during stage differentiation from the mammalian-infective to the insect form19. This study revealed many previously unknown components of the differentiation machinery that are involved in essential biological processes such as signaling, post-translational protein modifications, trafficking and nuclear transport.
Large-scale proteomic studies are extremely useful to approach cellular changes in a systematic way. However, to fully understand the functions and consequences of differential gene expression, detailed work on a molecular level is necessary. We therefore decided to specifically characterize putative proteins of unknown function that are highly upregulated in the BSF to learn more about novel trypanosomal adaptation factors for the mammalian host. We therefore focused on a well-investigated structure that is highly adapted to different host environments: the trypanosome flagellum.
Trypanosomes have a single flagellum attached lengthwise along the cell body. The flagellum contains a canonical eukaryotic axoneme and a paracrystalline accessory structure termed the paraflagellar rod (PFR)20,21. The flagellum emerges from the flagellar pocket (FP), an invagination of the cellular membrane and the exclusive site of exo- and endocytosis22,23. The axoneme is nucleated by a barrel-like microtubule structure called the basal body, which abuts the FP membrane, while the PFR is initiated once the flagellum has exited the FP24,25. The flagellum is attached to the body of the cell by a flagellar attachment zone (FAZ). The FAZ is a tripartite, trans-membrane adhesion complex that links the axoneme and PFR to two structures within the cell that run underneath the attached flagellum26,27,28,29. These structures are the FAZ filament and a specialized microtubule quartet. The composition of the flagellum has been investigated in several proteomic studies in PCFs30.
A growing body of evidence indicates that the flagellum is a major communication hub with the host environment, providing sensing and response to extracellular signals. A combination of flagellum purification together with affinity purification of surface-exposed proteins identified flagellum matrix and surface proteins in the BSF and gave an insight into flagellum signaling31. Stage-specific expression of individual paralogs within gene families was demonstrated by comparison of PCF and BSF cell surface proteomes, showing that the parasite surface is remodeled to allow adaptation to the different host environments32. The flagellar membrane is in direct contact with the outer environment. In the epimastigote form of the parasite, found in the tsetse fly, it forms branched flagellar outgrowths that are attached to the salivary gland epithelium33. PCF stage-specific flagellar surface receptor adenylate cyclases have been shown to localize specifically to the flagellum tip. This supports subdomain organization of the flagellar membrane and a microdomain model for flagellar cyclic AMP (cAMP) signaling in T. brucei34. Several other signaling molecules, virulence factors or potential motility factors from the flagellum such as BSF expression site associated gene ESAG4, metacaspase 4, glycosylphosphatidylinositol-phospholipase C and calflagins were reviewed previously34,35. Arginine kinase 3 (AK3) is highly expressed in PCF compared to BSF and was shown to confer advantage to the parasites during infection in tsetse flies36. Interestingly, AK3 shares a similarity with the flagellar targeting sequence of calflagins from T. brucei and flagellar calcium binding protein (FCaBP) from T. cruzi36. Recently, a study of several trypanosome species suggested that flagellar motion and swimming behavior restricts the parasite to distinguishable anatomic niches within the mammalian hosts37. Taken together, it is critical to learn more about flagellar composition and function to fully understand how these parasites adapt to different environments during their complicated life cycle.
Here we describe a putative flagellar component, Tb927.11.2400, which was discovered in our comparative proteome study16. Database searches revealed that Tb927.11.2400 shares 38% amino acid identity with PCF-specific TbFlabarin (Tb927.11. 2410). Flabarin was initially described in Leishmania donovani as a flagellar BAR domain protein38. LdFlabarin is targeted to the flagellum by a potential N-terminal acylation site and has a central BAR domain for membrane association and a C-terminal domain needed for the flagellar specificity. It forms a helicoidal structure from the base to the tip of the flagellum38. In vitro experiments suggest a morphogenetic and structural function since recombinant LdFlabarin associates with liposomes and triggers tubule formation38. We named Tb927.11.2400 TbFlabarin-Like (TbFlabarinL) following the convention adopted for Flabarins38. The two homologues in trypanosomes are likely a result of a gene duplication event. TbFlabarinL is downregulated 24 hours post induction of differentiation and undetectable in PCF. In contrast, TbFlabarin is upregulated very early during the differentiation process (2 h after induction) and is fully expressed 24 hours post induction. Protein expression levels of TbFlabarinL are 20-fold higher in BSF compared to PCF, which suggest that this protein might be a potential mammalian host-specific adaptation factor. We showed that TbFlabarinL is not essential for the growth of the parasite under cell culture conditions and that it is dispensable for developmental differentiation of trypanosomes. We generated a TbFlabarinL specific antibody and show that the endogenous protein localizes to the flagellum. We found that TbFlabarinL associates with both the flagellar membrane as well as with the PFR.
Materials and Methods
Computational analyses
A BLAST search with TbFlabarinL (Tb927.11.2400) was performed against the TriTryp database39. Alignments were obtained from constraint-based multiple alignment tool (COBALT)40 at the NCBI database and edited in Espript 3.041. Protein structures of TbFlabarinL and TbFlabarin were predicted by the Phyre2 server42 and edited in UCSF Chimera 1.10.243. Datasets for phylogenetic analyses were received from publicly available sources (Supplementary Table 1) for both Flabarin and FlabarinL genes, using BLASTP at an E-value cut-off of 10–20. All amino acid sequences were searched for conserved domains by Pfam. The datasets were aligned by MUSCLE and relevant positions were selected using Gblocks. Phylogenetic model selection with Modelgenerator favored LG + GAMMA model and ML trees were constructed using RAxML 8.1.17 with 1 000 bootstrap replicates. Bayesian Monte Carlo Markov (MCM) chain analysis was performed with GTR + GAMMA + CAT model using Phylobayes 3.3f running 8 independent chains for 10,000 cycles. Convergence of chains was estimated by comparison of bipartition frequencies in individual chains, discarding first 2,000 cycles.
Trypanosome cell lines and cultivation
Monomorphic BSF trypanosome “Single Marker” (SM) and 2T1 are both derivatives of Lister strain 427, antigenic type MITat 1.2, clone 221a (Doyle et al., 1980) and express T7 polymerase and Tetracycline repressor44. Monomorphic BSF trypanosomes were maintained in HMI-9 medium with 10% fetal calf serum (Sigma) and 5% CO245. PCF trypanosomes were cultured in modified SDM-79 medium with 10% fetal calf serum (Sigma) and 5% CO246. BSF and PCF cell densities were determined using a Coulter Counter Z2 (Beckman Coulter) particle counter, and cultures were diluted to maintain the cells in mid log growth phase. Transfection of monomorphic trypanosomes was performed as described previously47. Pleomorphic BSF trypanosomes AnTat1.1 were cultured in HMI-9 medium containing 1.1% methylcellulose. Cell density was determined using a Neubauer counting chamber (Brand) and cultures were diluted to maintain the cells in mid log growth phase. Cells were transfected as described previously48,49.
Differentiation of trypanosomes
Pleomorphic AnTat1.1 BSF were grown to a density of 2.5 × 106 cells/ml, diluted 1:5 in TDB (5 mM KCl, 80 mM NaCl, 1 mM MgSO4, 20 mM Na2HPO4, 2 mM NaH2PO4, 20 mM glucose, pH 7.4), filtered, sedimented by centrifugation (1500 × g, 10 min, 37 °C) and resuspended to a cell density of 2 × 106 cells/ml in DTM medium50. A mixture of cis-aconitate and isocitrate (3 mM) was added to induce differentiation and cells were incubated at 27 °C with 5% CO2 as described previously50.
Generation of transgenic trypanosome cell lines
To generate the TbFlabarinLRNAi cell line, a DNA fragment (positions 30–544) was amplified from genomic DNA using a forward primer containing SmaI and XhoI restriction sites and a reverse primer containing BamHI and XbaI sites. Sense and antisense fragments of the RNAi hairpin were ligated into the pRPAiSL vector51. Prior to transfection of 2T1 cells, the plasmids were linearized using AscI. Clones with a correctly integrated construct were selected as described previously51. RNAi was induced by addition of 1 μg/ml tetracycline to the cell culture and refreshed daily.
A PCR-based gene deletion approach was used to sequentially replace both alleles of TbFlabarinL with puromycin N-acetyl-transferase (PUR) and hygromycin phosphotransferase (HYG) open reading frames (ORFs) in MiTat1.2 SM. The ends of the TbFlabarinL 5′ UTR (59 nt) and 3′ UTR (60 nt) were flanked by sequences to amplify the HYG and PUR ORFs from the pHD309 HYG/PUR plasmid (gift from G.A.M. Cross). Cells were electroporated with the purified PCR products. The same constructs were used to delete both TbFlabarinL alleles in AnTat1.1 and AnTat1.1E-SmOx cells. Correct integration of the constructs was verified by PCR using primers binding in the 5′ and 3′ UTR, 5′ and 3′ ORF and within the puromycin and hygromycin resistance ORFs (Supplementary Figs S3 and S4). PCR was performed according to the manufacturer’s instructions of the Phusion Human Specimen Direct PCR Kit (Thermo Fisher Scientific).
TbFlabarinL was epitope-tagged at the N- or C-terminus in the endogenous locus as described in ref. 52. The plasmids for tagging were kindly provided by S. Kramer, University of Würzburg. Briefly, the TbFlabarinL ORF fragment without stop codon was amplified from genomic DNA and cloned into p3077_PAC_4xTY plasmid. The vector was linearized within the ORF fragment by HpaI prior to transfection of the cells. To tag TbFlabarinL at the C-terminus, a part of the TbFlabarinL ORF (positions 2-653) was amplified from genomic DNA and cloned into p3074_4xTY_BLE plasmid and linearized by HpaI before transfection of the cells. The same strategy was used to tag TbFlabarin (Tb927.11.2410) at the C-terminus. A fragment of the TbFlabarin ORF (positions 1-666) was cloned into p3074_4xTY_BLE plasmid and linearized with BclI enzyme prior transfection. To tag PAR1 (Tb927.11.13500) at the N-terminus, a fragment (positions 4-823) of the PAR1 ORF was cloned into the p3077_PAC_4xTY plasmid and EcoRI was used for linearization.
To express TbFlabarinL in PCF ectopically, its ORF was amplified from genomic DNA and cloned into pLEW100v5b2x_Phleo (gift from G.A.M. Cross) using XhoI and HindIII restriction sites. For ectopic expression of TbFlabarin in BSF, the TbFlabarin ORF was amplified from genomic DNA using a C-terminal primer containing a glycine-alanine-glycine (Gly-Ala-Gly) linker sequence followed by a Ty1-tag sequence and cloned into pLEW100v5b2x_Phleo using XhoI and HindIII restriction sites. Constructs were linearized with NotI for integration into the ribosomal spacer locus. Expression was induced by addition of 1 μg/ml tetracycline to the cell culture.
Polyclonal Antibody Generation
The TbFlabarinL ORF was amplified from genomic DNA of T. brucei using a primer containing a 10x His-tag sequence and a Gly-Ala-Gly linker sequence and cloned into the pETDUET-1 vector (Novagen) using NcoI and XhoI restriction sites. TbFlabarinL was expressed in Rosetta Blue Escherichia coli according to the manufacturer’s instructions and purified using an Äkta FPLC system with HisTrap FF crude 1 ml columns. Fractions containing recombinant TbFlabarinL were pooled, dialyzed in phosphate buffered saline and concentrated using a centrifugal filter unit with a 10 kDa cut off (Amicon). 1 mg of purified protein was sent to Pineda (Berlin) for antibody production. TbFlabarinL-specific antibody was affinity purified from the rabbit immune sera using the SulfoLink kit (Thermo Fisher Scientific) and recombinant TbFlabarinL according to the manufacturer’s instructions. Antibody specificity was tested in immunoblots using recombinant TbFlabarinL and whole-cell lysates of wild-type, ∆TbFlabarinL and TbFlabarinLRNAi cell lines.
All primer sequences used in this study are available upon request.
Western Blot (WB)
Whole-cell lysates were separated by SDS-PAGE on 10% polyacrylamide gels and transferred onto a Immobilon® PVDF membrane (MERCK MILLIPORE). The membrane was blocked 1 h at RT in PBS-5% milk and subsequently incubated 1 h at RT with polyclonal anti-TbFlabarinL rabbit antibody diluted 1:500 in PBS-1% milk. Monoclonal anti-PFR mouse antibody L13D6, monoclonal anti-tubulin TAT1 mouse antibody and BB2 anti-Ty1 mouse antibody were gifts from K. Gull (University of Oxford) and described elsewhere53,54,55. Anti-TbMORN1 rabbit antibody was described previously56. IRDye680- and IRDye800-coupled anti-mouse or anti-rabbit secondary antibodies were purchased from LI-COR Bioscience to detect the respective proteins with an Odyssey infrared imaging system (LICOR Bioscience).
Immunofluorescence (IF)
1 × 107 cells were harvested by centrifugation (1500 × g, 10 min, RT) and resuspended in 1 ml TDB. Cells were fixed in 2% PFA (10 min, RT). After 3 wash steps in PBS, cells were settled onto Poly-L-lysine-coated slides (20 min, RT) and permeabilized using 0.2% IGEPAL in PBS (5 min, RT). Non-specific epitopes were blocked with 1% BSA in PBS (1 hr at 37 °C). Cells were incubated with the primary TbFlabarinL antibody diluted 1:200 in PBS-0.1% BSA and/or monoclonal BB2 anti-Ty1 mouse antibody diluted 1:500 in PBS-0.1% BSA (1 hr, RT). After washing with PBS, the secondary antibody was applied (Alexa 594 anti-mouse, Alexa 488 anti-rabbit (Life Technologies)) in PBS-0.1% BSA (30 min, RT). DNA was stained with 1 μg/ml 4,6-diamidino-2-phenylindole (DAPI) and cells were embedded in ProLong Gold Antifade (Molecular Probes) and imaged on Leica DMI 6000B microscope. Images were processed using Huygens Essential XII deconvolution software (Scientific Volume Imaging).
Co-immunoprecipitation
Polyclonal TbFlabarinL antibody was immobilized on protein G Sepharose Fast Flow beads (GE Healthcare). Non-specific binding was blocked (1 hr, 4 °C) with 0.5% BSA in 20 mM sodium phosphate buffer pH 7.0. Four biological replicates per cell line (2 × 108 cells each) were harvested by centrifugation (1500 × g, 10 min, 4 °C) and washed in TDB buffer. Cells were lysed in 400 μl IP buffer (150 mM NaCl, 20 mM Tris-HCl pH 8, 10 mM MgCl2, 0.25% IGEPAL 1 mM DTT, Protease Inhibitors Cocktail without EDTA (Roche)) by sonication using a Bioruptor (Diagenode) with a 3 cycles 30 s high power pulse with 30 s pause setting. Cell lysates were incubated with the blocked beads (orbital mixing, overnight, 4 °C). Beads were washed in IP buffer. To elute the proteins, beads were resuspended in 65 μl NuPAGE LDS Sample Buffer (Novex, Life Technologies) supplemented with 100 mM DTT and incubated 10 min at 70 °C. The eluates were analyzed by mass spectrometry.
Mass Spectrometry
Protein samples were separated on a 4–12% NuPAGE Gel (Life Technologies) and stained with Coomassie colloidal blue (Life Technologies). The lanes were sliced and prepared by in-gel digestion with trypsin57. The peptides were stored on StageTips. Digested peptides were separated on a C18 reverse phase column (packed in-house, 20 cm, 75 μm inner diameter; packed with reprosilPur-1.8 [Dr. Maisch]) with a 105-minute gradient from 5 to 95 percent ACN on an Easy-nLC 1000. The solution was directly sprayed at 2.4 kV. The Q Exactive Plus was operated in a data-dependent Top10 acquisition mode with one full scan (70,000 resolution, max injection 20 ms, 300–1650 m/z) and up to 10 HCD fragment scans (17,500 resolution, max injection 120 ms). The raw spectra were analysed with MaxQuant ver1.5.1.058 using standard settings (except activated LFQ quantitation and match between runs) and the Trypanosoma brucei TriTrypDB-8.0_TbruceiTREU927 database. The protein Groups file was filtered to exclude contaminants, reverse entries and proteins only identified by site prior to statistical analysis. To obtain enrichment the LFQ values were log2 transformed and the mean between bait and control was calculated. To assess the statistical significance of the enrichment a Welch t-test for LFQ values between bait and control set was performed. Both values were visualized in a volcano plot using the ggplot2 package in R.
Mouse experiments
Animal experiments were performed according to EU regulations and approved by the Animal Ethics Committee of Instituto de Medicina Molecular (AEC_2011_006_LF_TBrucei_IMM). Male C57BL/6 wild-type mice (Charles River) were housed in the pathogen-free mouse facility of the Instituto de Medicina Molecular (IMM) with a 12:12-h light:dark cycle. 6 weeks-old mice were used for intraperitoneal infection with 2 × 103 parasites each. 5 mice were infected with T. brucei AnTat1.1E-SmOx parasites and 6 mice with the ∆TbFlabarinL cell line. Parasitemia was measured after infection by collecting blood from the tail vein. The vein was punctured with a gauge needle and 1 μl of blood was collected and diluted in 149 μl HMI-11. Parasites numbers were manually quantified using a Neubauer counting chamber. The minimum of detectable parasites in blood is 1.5 × 105 cells/ml.
Isolation of cytoskeletons and flagella
Cytoskeleton- and flagella-enriched fractions were prepared following a published protocol59,60 with minor modifications. Briefly, 1×108 cells were harvested by centrifugation (1500 × g, 10 min, 4 °C) and washed in 1 ml PEME buffer (2 mM EGTA, 1 mM MgSO4, 0.1 mM EDTA, 0.1 M piperazine-N,N′-bis (2-ethanesulfonic acid)-NaOH (PIPES-NaOH), pH 6.9). Cytoskeletons (P1) were obtained by extracting the cells in PEME buffer with 1% IGEPAL (15 min, RT, orbital mixing) and separated from the detergent soluble supernatant (S1) by centrifugation (3400 × g, 5 min, 4 °C). Flagella were isolated from the extracted cytoskeletons (P1) by incubation in PEME buffer with 1% IGEPAL and 1M KCl (30 min, RT, orbital mixing), which depolymerized the corset microtubules, followed by centrifugation (6000 × g, 15 min, 4 °C) to separate S2 and P2 fractions. All solutions were supplemented with Complete protease inhibitors cocktail without EDTA (Roche).
Results
TbFlabarinL originated from a gene duplication event
TbFlabarinL (Tb927.11. 2400) was identified in a proteomic study16 as a BSF-specific, putative protein of unknown function. A BLAST search in the TriTryp database39 revealed a 38% amino acid (AA) identity and 60% similarity61,62 to the product of the neighboring Tb927.11.2410 gene (Fig. 1A). Tb927.11.2410 is TbFlabarin, a flagellar BAR domain protein in T. brucei. Flabarins were first described in Leishmania spp38. TbFlabarin consists almost entirely of a BAR/IMD-like domain, which extends over the region of 8–212 AA out of a total 222 AA length of the protein. Despite the high similarity to TbFlabarin (60% AA similarity), a BAR/IMD like domain was not detected by the prediction software in TbFlabarinL. However, predicted structural models42 of both TbFlabarin and TbFlabarinL feature a triple helix coiled coil architecture (Fig. 1B). Such a structure is characteristic of BAR proteins and enables formation of banana-shaped homo- or heterodimers known for their membrane curvature generation (reviewed in ref. 63).

(A) Alignment of TbFlabarinL (Tb927.11.2400) with TbFlabarin (Tb927.11.2410). A BLAST search with the Tb927.11.2400 protein sequence identified a Trypanosoma brucei flabarin homologue (Tb927.11.2410). The amino acid sequences of TbFlabarinL and TbFlabarin have 38% identity (indicated by black boxes) and 60% similarity (indicated by white boxes). (B) Sequence-based structure modeling of TbFlabarinL and TbFlabarin. An overlay of the two structures is shown.
TbFlabarin and TbFlabarinL are both found on chromosome 11 separated by an ~12 kb stretch of non-coding sequence. The TbFlabarin coding sequence (CDS) is on the complementary DNA strand. Orthologs of TbFlabarinL are present within the orthology group OG5_185161 from Ortho MCL DB64,65 in T. brucei gambiense, T. vivax and T. evansi. In contrast to Flabarin OG5_148786, FlabarinL is neither present in the genomes of sequenced monoxenous (one-host) trypanosomatids, nor in the genus Leishmania and American trypanosomes. Phylogenetic analysis revealed a gene duplication event in African trypanosomes that gave rise to the FlabarinL gene (Fig. 2). Interestingly, FlabarinL is missing in T. congolense and T. equiperdum, which might be due to incompleteness of these genomes. On the other hand, it could reflect specific host adaptation requirements.

Phylogenetic analysis of Flabarins across Trypanosomatida.
A maximum likelihood phylogenetic tree based on alignments of Flabarin and FlabarinL proteins. The scale bar indicates the inferred number of amino acid substitutions per site. The red dot indicates a gene duplication event that gave rise to the FlabarinL gene, which is present only in a subset of African trypanosomes.
To confirm the expression profiles of the mass spectrometry study, we generated a TbFlabarinL-specific antibody. Recombinant full-length TbFlabarinL was purified from E. coli (Supplementary Fig. S1) and used for immunization of a rabbit. TbFlabarinL-specific antibody was affinity purified from the immune sera and used in Western blot (WB) analysis (Fig. 3). A band of the expected molecular weight could be detected in BSF whole cell lysates but not in lysates from PCF, supporting the conclusion from mass-spectrometry analysis that TbFlabarinL is a BSF stage-specific protein.

TbFlabarinL is a BSF stage-specific protein.
Immunoblot analysis of whole cell lysates from PCF and BSF cells using affinity purified anti-TbFlabarinL antibodies. Expression of TbFlabarinL was detected in BSF cells only. Immunoblotting with anti-Histone H3 served as a loading control. A representative blot of multiple (N > 3) independent experiments is shown. Re: recombinant His-tagged TbFlabarinL.
TbFlabarinL localizes to the flagellum in BSF trypanosomes
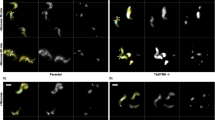
TbFlabarinL-specific antibody was used in immunostaining to determine the subcellular localization of TbFlabarinL in BSF trypanosomes. A ∆TbFlabarinL cell line (described in later sections) and PCF cells were used as controls. Cells were stained with TbFlabarinL-specific antibody and TAT1 anti-tubulin antibody (Fig. 4). TbFlabarinL localized to the flagellum in BSFs, with a labeling pattern that initiated close to the kinetoplast (small spot in DAPI channel) and extended lengthwise along the cell body. No signal was detected from the ∆TbFlabarinL cells, confirming the specificity of the labeling pattern. No signal was detected in PCF cells, further confirming the stage-specific expression of the TbFlabarinL protein.

TbFlabarinL is localized to the flagellum.
Immunofluorescence analysis using the anti-TbFlabarinL antibody showed that TbFlabarinL localized to the flagellum of wild-type (WT) T. brucei BSF cells. No labeling pattern was observed in the BSF knockout cell line (BSF ΔTbFlabarin L) or PCF WT cells Scale bar 3 μm.
We observed that N-terminal tagging of TbFlabarinL resulted in a mislocalization of TbFlabarinL. Cells in which the ORF of only one allele was fused to a sequence that encodes for a 4xTy1 epitope tag by homologous recombination were stained with BB2 anti-Ty1 antibody and TbFlabarinL specific antibodies. The tagged version of TbFlabarinL remained in the cytoplasm (Fig. 5A), whereas the wild-type version of could be detected in the flagellum. Interestingly, the first 24 N-terminal AA of TbFlabarinL share 75% identity with the N-terminal sequence of flagellar calcium binding protein (FCaBP) of T. cruzi (Fig. 5B). The first 24 AA are responsible for flagellar localization and for the association of FCaBP with the inner leaflet of the flagellar membrane66. These effects are mediated by the presence of a myristoylated glycine in position 2, palmitoylated cysteine in position 4, and two conserved lysines, TbFlabarinL contains glycine in position 2 and cysteine in position 3 and two positively charged lysines. The similarity of the N-termini of TbFlabarinL and FCaBP and mislocalization of TbFlabarinL after in-situ N-terminal tagging implies TbFlabarinL flagellar membrane localization is probably mediated by similar targeting mechanisms.

In situ N-terminal tagging results in mislocalization of TbFlabarinL to the cytoplasm.
(A) The N-terminally Ty1-tagged TbFlabarinL (red) shows localization in the cytosol whereas the TbFlabarinL WT protein (green) also localizes to the flagellum. The TbFlabarinL-specific antibody recognizes both the WT and the tagged allele of TbFlabarinL. Scale bar 3 μm. (B) The N-terminus of TbFlabarinL is similar to N-terminal sequences of Flagellar Calcium Binding Protein (FCaBP) of T. cruzi. Elements potentially necessary for the association with the flagellar membrane are highlighted. Palmitoylated and myristoylated amino acids are marked in red and blue, respectively. Conserved lysines are marked in yellow. Identical and conserved residues are boxed in black and white respectively.
To analyze the similarity of TbFlabarinL and TbFlabarin, and to test if a common protein targeting mechanism apply for the procyclic protein, C-terminally Ty1 epitope-tagged TbFlabarin was ectopically expressed in BSF. TbFlabarin localized clearly to the flagellum with a discontinuous pattern similar to that of TbFlabarinL. Staining with anti-Ty1 and anti-TbFlabarinL antibodies showed an overlap of the signal in distinct regions of the flagellum (Fig. 6A). Conversely, ectopically overexpressed TbFlabarinL in PCF localized to the flagellum in a more continuous pattern along the whole length of the flagellum (Fig. 6B) presumably due to high expression levels (200-fold overexpression compared to endogenous protein levels in PCF). In situ C-terminally Ty1-tagged TbFlabarin in PCF localized to the flagellum and displayed a patchy pattern similar to that of TbFlabarinL in BSF (Fig. 6C). These observations are consistent with the hypothesis that TbFlabarinL and TbFlabarin have a similar stage-specific function in the flagellum of T. brucei.

TbFlabarin and TbFlabarinL localization in the BSF and PCF T. brucei.
Immunofluorescence staining using anti-Ty1 (red) and anti-TbFlabarinL (green) antibodies showed that ectopically expressed TbFlabarin in BSF had a similar patchy localization pattern as TbFlabarinL. The localization of the two proteins overlaps in some regions. (B) Ectopically expressed TbFlabarinL localized to the flagellum in PCF. (C) In situ C-terminally tagged TbFlabarin in PCF showed a similar localization pattern in the flagellum to the one of TbFlabarinL in BSF. Scale bar 3 μm.
TbFlabarinL is associated with the paraflagellar rod and the flagellar membrane
To learn more about the function of TbFlabarinL, we employed a co-immunoprecipitation (IP) approach to find interacting partners of TbFlabarinL. Four biological replicates of WT and ∆TbFlabarinL as a control were used in IP and analyzed by mass spectrometry. Two putative interacting partners Tb927.11.3840 and Tb927.11.13500 (Fig. 7A) were found. The first candidate interaction partner is an unknown putative protein with no conserved domains. The second candidate interaction partner is PAR1, a previously-described paraflagellar rod component67,68,69. To verify the result of the IP, an endogenous replacement cell line was generated by homologous recombination, with the PAR1 ORF fused to a sequence that encodes for a 4xTy1 epitope tag. These cells were stained with the anti-Ty1 antibody for immunolocalization studies. As expected, 4xTy1-PAR1 localized to the flagellum (Fig. 7B). Detection with anti-Ty1 and TbFlabarinL specific antibodies showed a partial overlap of TbFlabarinL and Ty1-PAR1 in distinct parts of the flagellum, which supported the result of the IP. Interestingly, TbFlabarinL localization revealed a different pattern compared to PAR1. TbFlabarinL showed an interrupted and punctate pattern throughout the flagellum (Fig. 7B).

Immunoprecipitation using TbFlabarinL specific antibody.
(A) Volcano plot of co-enriched proteins after anti-TbFlabarinL IP obtained by label-free quantitative mass spectrometry of four parallel biological replicates. Besides TbFlabarinL (Tb927.11.2400), two other proteins were enriched: Tb927.11.3840 and Tb927.11.13500. Tb927.11.3840 is an uncharacterized hypothetical protein. Tb927.11.13500 is PAR1, a paraflagellar rod component. (B) TbFlabarinL partially overlaps with the PFR component Par1 in the flagellum. Immunofluorescence analysis using the TbFlabarinL specific antibody showed that TbFlabarinL localized to the flagellum of T. brucei. A co-staining of the PFR component PAR1 (Ty1-tagged, red) and anti-TbFlabarinL (green) antibody showed TbFlabarinL and PAR1 overlap in certain regions of the flagellum. Scale bar 3 μm.
The results of the IP experiment as well as the partial overlap of PAR1 and TbFlabarinL immunostaining imply a possible association of TbFlabarinL with the PFR structure of the flagellum. In contrast, the N-terminal sequence of TbFlabarinL seems to attach TbFlabarinL to the flagellar membrane. To probe the membrane association of TbFlabarinL, a biochemical cell fractionation was performed. In the first step, the detergent soluble fraction S1 (cytoplasm/membranes) was separated from P1 (cytoskeleton). In the second step, the cytoskeletal P1 fraction was treated with 1 M KCl to depolymerize the microtubule corset and the FAZ filament and thus separate S2 (corset microtubules together with FAZ) from P2 (PFR, axoneme, basal body and flagellar pocket collar). WB analysis of equal fractions (10% of input material) revealed that TbFlabarinL is found not only within the soluble cytoplasmic and membrane fractions but also within the cytoskeletal fractions both before and after separation of the flagella from the cytoskeleton (Fig. 8). This result supports the observations described in previous experiments, which suggested a dual interaction with the flagellar membrane as well as with structural components of the flagellum such as the PFR.

Biochemical cell fractionation.
(A) Fractionation scheme: The input sample (I) was taken after non-ionic detergent extraction of BSF cells. The cells were then separated by centrifugation into a detergent-soluble supernatant (S1) and detergent-insoluble cytoskeletal pellet (P1). The P1 fraction was solubilized in high salt to depolymerize corset microtubules and further separated by centrifugation into supernatant (S2) and pellet (P2) fractions. (B) Fractionation of TbFlabarinL. Equal fractions (10%) were loaded on a gel and analyzed by immunoblotting. Anti-PFR, anti-tubulin and TbFlabarinL specific antibodies were used. TbFlabarinL was found in all fractions.
TbFlabarinL is not essential for BSF in vitro and in vivo
In order to examine the effect of TbFlabarinL depletion on the viability of T. brucei, an inducible RNAi knockdown cell line was generated. TbFlabarinL expression was significantly reduced 24 h after induction and almost below detection level after 48 h (Fig. 9A). The growth of the parasites upon induction of RNAi was monitored. There was no growth phenotype observed upon depletion of TbFlabarinL in three independent clones (Fig. 9B and Supplementary Fig. S2), indicating that TbFlabarinL is not an essential gene for the survival of the parasite under cell culture conditions. To further pursue this indication, a ∆TbFlabarinL knockout cell line was generated in BSF trypanosomes by homologous recombination. The deletion of both TbFlabarinL alleles was verified by integration PCR (Supplementary Fig. S3) and by WB analysis (Fig. 9C). No difference was observed in growth between the parental and the ∆TbFlabarinL cell lines (Fig. 9D). To test the role of TbFlabarinL in vivo, we infected mice with WT and ∆TbFlabarinL pleomorphic cell lines. We detected no significant differences between the parasitemia profiles and survival curves, suggesting that deletion of TbFlabarinL has no effect on mammalian infections (Fig. S6). We concluded that TbFlabarinL is not an essential gene for the viability of BSF T. brucei under cell culture conditions and in vivo.

Depletion of TbFlabarinL in BSF T. brucei.
(A) TbFlabarinL protein was almost undetectable 48 h after induction of RNAi, as determined by anti-TbFlabarinL immunoblot. A weak additional band with the size of 25 kDa is detectable after RNAi induction. PFR was used as a loading control. (B) Depletion of TbFlabarinL by RNAi had no effect on the growth of BSF T. brucei under cell culture conditions. A cumulative growth curve of one representative clone is shown. Two independent clones showed a similar phenotype (see Supplementary Fig. S2). (C) An immunoblot analysis using TbFlabarinL specific antibody confirmed deletion of TbFlabarinL in two independent clones (c1 and c2) and a non-clonal population (pool). A weak additional band with the size of 25 kDa is detectable after deletion of TbFlabarinL. Immunoblotting with anti-PFR antibodies was used as a loading control. (D) The ∆TbFlabarinL cells showed no growth defect compared to the wild-type controls (N = 3).
TbFlabarinL is dispensable for developmental differentiation
TbFlabarinL has a very interesting expression profile during developmental differentiation. It is highly upregulated in LS and SS forms, still detectable during early differentiation but is downregulated below detection levels 48 h after induction of differentiation19. To test if TbFlabarinL could be involved in BSF to PCF transition, a stable ∆TbFlabarinL cell line was generated in a pleomorphic T. brucei strain. So-called monomorphic parasites, which we used in the previous experiments, are culture adapted and very convenient for reverse genetics, but initiation of differentiation is inherently inefficient and asynchronous. In pleomorphic field strains of T. brucei, differentiation is very efficient and both steps of the differentiation process (LS to SS and SS to PCF) can be monitored in cell culture. Furthermore, pleomorphic strains still respond to stumpy induction factor (SIF) with growth arrest in vivo and are therefore the better system to study differentiation cell biology and virulence in vivo. Hence, we generated pleomorphic ∆TbFlabarinL trypanosomes in strain AnTat1.1 as described above. The loss of both TbFlabarinL alleles was verified by integration PCR (Supplementary Fig. S4) and WB analysis (Fig. 10A). First, the growth of the ∆TbFlabarinL cell line was compared to the parental cell line and only a mild growth phenotype was observed (Fig. 10B), which is in agreement with the result seen in the monomorphic ∆TbFlabarinL cell line (Fig. 10). Second, the pleomorphic cells were grown to a high density to induce SS formation and then further differentiated for 52 hrs. WB analysis (Fig. 10C) confirmed the downregulation of TbFlabarinL during differentiation seen in proteomic analysis19. ∆TbFlabarinL and parental cells grew equally well during developmental differentiation (Fig. 10D), which suggests that TbFlabarinL is dispensable for this process. A second very faint band migrating slightly higher than TbFlabarinL was detected by anti-TbFlabarinL antibody in the ∆TbFlabarinL cell lines after induction of differentiation (Fig. 10C). The band is detectable in the WT pleomorphic cells during differentiation from SS to PCF at the time point 28 h post differentiation, when the TbFlabarinL band becomes weaker. The same band is detectable during the differentiation of ∆TbFlabarinL at the time point 52 h. To exclude that the anti-TbFlabarinL antibody can recognize TbFlabarin, WB analysis of a BSF cell line ectopically expressing Ty1 epitope-tagged TbFlabarin was performed (Supplementary Fig. S5). Both anti-TbFlabarinL and anti-Ty1 antibody could only detect one band, which supports the specificity of the TbFlabarinL-specific antibody.

TbFlabarinL deletion in pleomorphic BSF T. brucei.
(A) Immunoblot analysis with TbFlabarinL specific antibody verified the deletion of TbFlabarinL. An anti-PFR immunoblot was used as a loading control. (B) Deletion of TbFlabarinL causes a mild growth defect (N = 3). (C) Immunoblot analysis with TbFlabarinL specific antibody confirmed the downregulation of TbFlabarinL at different time points during differentiation from long slender (LS) to short stumpy (SS) and then 2, 28 and 52 hours after induction of differentiation. A weak additional band with the size of 25 kDa is detectable after induction of differentiation. (D) Pleomorphic trypanosomes were successfully differentiated from LS to PCF. There was no difference in the growth rate between the parental (WT) and ∆TbFlabarinL cell line during and after differentiation (N = 1).
In summary, TbFlabarinL is a result of a gene duplication of TbFlabarin, which occurred in African trypanosomes. We generated a TbFlabarinL antibody and showed that it localizes to the flagellum. The results of IP experiments and a biochemical cell fractionation suggest a dual interaction of TbFlabarinL with the flagellar membrane as well as with structural components of the flagellum such as the PFR. TbFlabarinL is not essential for the growth and differentiation of T. brucei under cell culture conditions.
Discussion
Trypanosomes must adapt to different host environments during their complex life cycle. Large-scale proteomic approaches provide information on changes at the cellular level in a systematic way. However, a detailed work on single components is necessary to understand the adaptation mechanisms on a molecular level. Here, we have performed a detailed characterization of a BSF stage-specific putative host adaptation factor Tb927.11.2400 identified in a SILAC-based comparative proteome study16. Tb927.11.2400 shares a 38% amino acid identity with TbFlabarin (Tb927.11.2410), a PCF stage-specific16 flagellar BAR domain protein. The BAR domain is missed by the prediction software in Tb927.11.2400 and this is why we named it TbFlabarin Like (TbFlabarinL).
Flabarin was first described in L. donovani as a flagellar BAR domain protein38. It forms a helical structure from the base to the tip of the flagellum38. In vitro experiments suggested a morphogenetic and structural function since recombinant LdFlabarin associates with liposomes and triggers tubule formation38. Recently, membranous nanotube formation was reported in T. brucei70. Those nanotubes appear to originate from the flagellar membrane and seem to dissociate into free extracellular vesicles (EVs). The protein composition of isolated EVs was determined by mass spectrometry and revealed the presence of TbFlabarinL together with 155 other proteins70. TbFlabarinL could be involved in nanotube/EVs formation or play a role in the fusion ability of the vesicles with other trypanosomes or erythrocytes or have an influence on the contents of the vesicles.
TbFlabarinL and TbFlabarin are both found adjacent on chromosome 11. We demonstrated that TbFlabarinL is a result of a gene duplication event that occurred in African trypanosomes. However, in T. congolense and T. equiperdum a secondary loss of FlabarinL seems to have happened. This could be explained by the unique life cycles of these two species. In contrast to T. brucei, T. congolense is a strictly intravascular parasite and does not traverse different tissues71. T. equiperdum is essentially a tissue parasite in the reproductive system of Equidae family animals with a very low parasitemia in the blood and is transmitted venereally72. A second reason for the absence of FlabarinL in T. congolense and T. equiperdum could be the limited sequencing data available for both species. While the BSF stage upregulation suggested an essential role of TbFlabarinL in the mammalian host, gene depletion and deletion experiments revealed that TbFlabarinL is not an essential gene for the growth and differentiation of trypanosomes in cell culture. Infections in mice indicated that TbFlabarinL is also dispensable in vivo. Based on the distribution of TbFlabarinL across the different trypanosomatida species, we speculate that TbFlabarinL was acquired as an adaptation to the lifestyle of different African trypanosomes, in particular to their ability to leave the bloodstream in the mammalian host and migrate through different tissues in the advanced stages of the infection. Alternatively, TbFlabarinL might be important for developmental differentiation in the Tsetse fly. Unfortunately, we do not have information about changes in expression levels of TbFlabarinL in different stages in the fly.
We generated a TbFlabarinL-specific antibody and could detect TbFlabarinL in the flagellum in a punctate pattern. Masking the N-terminal end of TbFlabarinL with an epitope tag resulted in mislocalization of the protein. This provided evidence for the importance of the N-terminal sequence in targeting TbFlabarinL to the flagellum. A closer inspection of the TbFlabarinL N-terminus allowed us to hypothesize that the presence of positively charged lysines and potentially myristoylated glycine and palmitoylated cysteine might be important for targeting TbFlabarinL to the flagellar membrane as shown previously for FCaBP in T. cruzi66. A recent study of the flagellar arginine kinase 3 (AK3) summarizes so far known proteins such as TbCaf17, TbCaf24, TbCaf44 which all share similar flagellar address with the FCaBP36. In addition, the BAR domain of LdFlabarin mediates its association with the flagellar membrane38. A structural model of both TbFlabarinL and TbFlabarin predicted a coiled-coil helical architecture identical with LdFlabarin despite a missing BAR domain per se and suggested a membrane association.
The punctate TbFlabarinL localization pattern could be explained by a potential association of the TbFlabarinL with discrete complexes in the flagellar membrane due to the presence of dual acylation. Dually acylated proteins have been shown to prefer association with membrane microdomains enriched in sphingolipids and cholesterol (reviewed in ref. 73). IP using anti-TbFlabarinL specific antibody identified PAR1 as a possible interacting partner of TbFlabarinL. PAR1 is a component of the PFR and unlike TbFlabarinL, it was detected in a comparative proteomic study74 together with 20 novel components of the PFR. Cell fractionation revealed that TbFlabarinL is present not only in the detergent-soluble fraction but also it is tightly associated with the isolated flagella and cytoskeletal fractions containing the PFR, which is in concordance with the predicted flagellar membrane localization as well as the result of the IP experiment. We suggest that the association of TbFlabarinL with the flagellar membrane might be only transient and function via a myristoyl switch mechanism, where the exposure of the myristate moiety could be ligand mediated as it is in the case of ADP ribosylation factor 1 (Arf1)75. The binding of Arf1 to the membrane is regulated by guanine nucleotide (GDP). When GDP is bound, the myristoylated N-terminal helix is sheltered in a hydrophobic groove of Arf1 and dissociates from the membrane76. Likewise when a ligand binds to TbFlabarinL, it might dissociate from the membrane and interact with the cytoskeletal components of the flagellum. While this manuscript was in preparation, Tetaud and colleagues published the characterization of TbFlabarin in PCF77. They showed that TbFlabarin mRNA is mainly expressed in PCF and that the flagellar localization of the protein is dependent on two cysteines at the N-terminus, which might mediate the strong association of the protein with the parasite’s membrane in their palmitoylated form.
In this study we present a characterization of two flabarin orthologs in T. brucei with a major focus on TbFlabarinL, which is BSF stage specific in contrast to PCF specific TbFlabarin. Further research is still needed to fully understand the function of flabarins in trypanosomes.
Additional Information
How to cite this article: Cicova, Z. et al. Two flagellar BAR domain proteins in Trypanosoma brucei with stage-specific regulation. Sci. Rep. 6, 35826; doi: 10.1038/srep35826 (2016).
References
Jensen, R. E. & Englund, P. T. Network news: the replication of kinetoplast DNA. Annu. Rev. Microbiol. 66, 473–491 (2012).
Lopes, A. H. et al. Trypanosomatids: Odd Organisms, Devastating Diseases. Open Parasitol. J. 4 (2010).
Trindade, S. et al. Trypanosoma brucei Parasites Occupy and Functionally Adapt to the Adipose Tissue in Mice. Cell Host Microbe 19, 837–848 (2016).
Morrison, L. J., Marcello, L. & McCulloch, R. Antigenic variation in the African trypanosome: molecular mechanisms and phenotypic complexity. Cell. Microbiol. 11, 1724–1734 (2009).
Vassella, E., Reuner, B., Yutzy, B. & Boshart, M. Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J. Cell Sci. 110, 2661–2671 (1997).
Roditi, I. et al. Procyclin gene expression and loss of the variant surface glycoprotein during differentiation of Trypanosoma brucei. J. Cell Biol. 108, 737–746 (1989).
Tetley, L. & Vickerman, K. Differentiation in Trypanosoma brucei: host-parasite cell junctions and their persistence during acquisition of the variable antigen coat. J. Cell Sci. 74, 1–19 (1985).
Vickerman, K. Developmental cycles and biology of pathogenic trypanosomes. Br. Med. Bull. 41, 105–114 (1985).
Siegel, T. N., Hekstra, D. R., Wang, X., Dewell, S. & Cross, G. A. M. Genome-wide analysis of mRNA abundance in two life-cycle stages of Trypanosoma brucei and identification of splicing and polyadenylation sites. Nucleic Acids Res. 38, 4946–4957 (2010).
Queiroz, R., Benz, C., Fellenberg, K., Hoheisel, J. D. & Clayton, C. Transcriptome analysis of differentiating trypanosomes reveals the existence of multiple post-transcriptional regulons. BMC Genomics 10, 495 (2009).
Kabani, S. et al. Genome-wide expression profiling of in vivo-derived bloodstream parasite stages and dynamic analysis of mRNA alterations during synchronous differentiation in Trypanosoma brucei. BMC Genomics 10, 427 (2009).
Clayton, C. The regulation of trypanosome gene expression by RNA-binding proteins. PLoS Pathog. 9, e1003680 (2013).
Manful, T., Fadda, A. & Clayton, C. The role of the 5′-3′ exoribonuclease XRNA in transcriptome-wide mRNA degradation. RNA 17, 2039–2047 (2011).
Vasquez, J.-J., Hon, C.-C., Vanselow, J. T., Schlosser, A. & Siegel, T. N. Comparative ribosome profiling reveals extensive translational complexity in different Trypanosoma brucei life cycle stages. Nucleic Acids Res. 42, 3623–3637 (2014).
Jensen, B. C. et al. Extensive stage-regulation of translation revealed by ribosome profiling of Trypanosoma brucei. BMC Genomics 15, 911 (2014).
Butter, F. et al. Comparative proteomics of two life cycle stages of stable isotope-labeled Trypanosoma brucei reveals novel components of the parasite’s host adaptation machinery. Molecular & Cellular Proteomics 12, 172–179 (2012).
Gunasekera, K., Wüthrich, D., Braga-Lagache, S., Heller, M. & Ochsenreiter, T. Proteome remodelling during development from blood to insect-form Trypanosoma brucei quantified by SILAC and mass spectrometry. BMC Genomics 13, 556 (2012).
Urbaniak, M. D., Guther, M. L. S. & Ferguson, M. A. J. Comparative SILAC proteomic analysis of Trypanosoma brucei bloodstream and procyclic lifecycle stages. PLoS One 7, e36619 (2012).
Dejung, M. et al. Quantitative Proteomics Uncovers Novel Factors Involved in Developmental Differentiation of Trypanosoma brucei. PLoS Pathog. 12, e1005439 (2016).
Wheeler, R. J., Gluenz, E. & Gull, K. The limits on trypanosomatid morphological diversity. PLoS One 8, e79581 (2013).
Gull, K. The cytoskeleton of trypanosomatid parasites. Annu. Rev. Microbiol. 53, 629–655 (1999).
Engstler, M. et al. Kinetics of endocytosis and recycling of the GPI-anchored variant surface glycoprotein in Trypanosoma brucei. J. Cell Sci. 117, 1105–1115 (2004).
Grünfelder, C. G. et al. Endocytosis of a glycosylphosphatidylinositol-anchored protein via clathrin-coated vesicles, sorting by default in endosomes, and exocytosis via RAB11-positive carriers. Mol. Biol. Cell 14, 2029–2040 (2003).
Lacomble, S. et al. Basal body movements orchestrate membrane organelle division and cell morphogenesis in Trypanosoma brucei. J. Cell Sci. 123, 2884–2891 (2010).
Lacomble, S. et al. Three-dimensional cellular architecture of the flagellar pocket and associated cytoskeleton in trypanosomes revealed by electron microscope tomography. J. Cell Sci. 122, 1081–1090 (2009).
Vaughan, S., Kohl, L., Ngai, I., Wheeler, R. J. & Gull, K. A repetitive protein essential for the flagellum attachment zone filament structure and function in Trypanosoma brucei. Protist 159, 127–136 (2008).
Sunter, J. D., Varga, V., Dean, S. & Gull, K. A dynamic coordination of flagellum and cytoplasmic cytoskeleton assembly specifies cell morphogenesis in trypanosomes. J. Cell Sci. 128, 1580–1594 (2015).
Zhou, Q., Hu, H., He, C. Y. & Li, Z. Assembly and maintenance of the flagellum attachment zone filament in Trypanosoma brucei. J. Cell Sci. 128, 2361–2372 (2015).
Sunter, J. D. & Gull, K. The Flagellum Attachment Zone: ‘The Cellular Ruler’ of Trypanosome Morphology. Trends Parasitol. 32, 309–324 (2016).
Subota, I. et al. Proteomic analysis of intact flagella of procyclic Trypanosoma brucei cells identifies novel flagellar proteins with unique sub-localization and dynamics. Mol. Cell. Proteomics 13, 1769–1786 (2014).
Oberholzer, M. et al. Independent analysis of the flagellum surface and matrix proteomes provides insight into flagellum signaling in mammalian-infectious Trypanosoma brucei. Mol. Cell. Proteomics 10, M111.010538 (2011).
Shimogawa, M. M. et al. Cell surface proteomics provides insight into stage-specific remodeling of the host-parasite interface in Trypanosoma brucei. Mol. Cell. Proteomics M114.045146 (2015).
Tetley, L. & Vickerman, K. Differentiation in Trypanosoma brucei: host-parasite cell junctions and their persistence during acquisition of the variable antigen coat. J. Cell Sci. 74, 1–19 (1985).
Saada, E. A. et al. Insect stage-specific receptor adenylate cyclases are localized to distinct subdomains of the Trypanosoma brucei Flagellar membrane. Eukaryot. Cell 13, 1064–1076 (2014).
Langousis, G. & Hill, K. L. Motility and more: the flagellum of Trypanosoma brucei. Nat. Rev. Microbiol. 12, 505–518 (2014).
Ooi, C.-P. et al. The Flagellar Arginine Kinase in Trypanosoma brucei Is Important for Infection in Tsetse Flies. PLoS One 10, e0133676 (2015).
Bargul, J. L. et al. Species-Specific Adaptations of Trypanosome Morphology and Motility to the Mammalian Host. PLoS Pathog. 12, e1005448 (2016).
Lefebvre, M. et al. LdFlabarin, a new BAR domain membrane protein of Leishmania flagellum. PLoS One 8, e76380 (2013).
Aslett, M. et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 38, D457–D462 (2010).
Papadopoulos, J. S. & Agarwala, R. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 23, 1073–1079 (2007).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Wirtz, E., Leal, S., Ochatt, C. & Cross, G. M. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99, 89–101 (1999).
Hirumi, H. & Hirumi, K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 75, 985–989 (1989).
Brun, R. & Schönenberger . Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Short communication. Acta Trop. 36, 289–292 (1979).
Burkard, G., Fragoso, C. M. & Roditi, I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 153, 220–223 (2007).
Vassella, E. et al. Deletion of a novel protein kinase with PX and FYVE-related domains increases the rate of differentiation of Trypanosoma brucei. Mol. Microbiol. 41, 33–46 (2001).
MacGregor, P., Rojas, F., Dean, S. & Matthews, K. R. Stable transformation of pleomorphic bloodstream form Trypanosoma brucei. Mol. Biochem. Parasitol. 190, 60–62 (2013).
Overath, P., Czichos, J. & Haas, C. The effect of citrate/ cis-aconitate on oxidative metabolism during transformation of Trypanosoma brucei. Eur. J. Biochem. 160, 175–182 (1986).
Alsford, S. & Horn, D. Single-locus targeting constructs for reliable regulated RNAi and transgene expression in Trypanosoma brucei. Mol. Biochem. Parasitol. 161, 76–79 (2008).
Kelly, S. et al. Functional genomics in Trypanosoma brucei: A collection of vectors for the expression of tagged proteins from endogenous and ectopic gene loci. Mol. Biochem. Parasitol. 154, 103–109 (2007).
Kohl, L., Sherwin, T. & Gull, K. Assembly of the paraflagellar rod and the flagellum attachment zone complex during the Trypanosoma brucei cell cycle. J. Eukaryot. Microbiol. 46, 105–109 (1999).
Woods, A. et al. Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies. J. Cell Sci. 93, 491–500 (1989).
Bastin, P., Bagherzadeh, A., Matthews, K. R. & Gull, K. A novel epitope tag system to study protein targeting and organelle biogenesis in Trypanosoma brucei. Mol. Biochem. Parasitol. 77, 235–239 (1996).
Morriswood, B. et al. Novel bilobe components in Trypanosoma brucei identified using proximity-dependent biotinylation. Eukaryot. Cell 12, 356–367 (2013).
Shevchenko, A., Tomas, H., Havlis, J., Olsen, J. V. & Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 (2006).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Esson, H. J. et al. Morphology of the trypanosome bilobe, a novel cytoskeletal structure. Eukaryot. Cell 11, 761–772 (2012).
Morriswood, B. & Schmidt, K. A MORN-repeat protein facilitates protein entry into the flagellar pocket of Trypanosoma brucei. Eukaryot. Cell EC. 00094-15 (2015).
Schäffer, A. A. et al. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 29, 2994–3005 (2001).
Altschul, S. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Mim, C. & Unger, V. M. Membrane curvature and its generation by BAR proteins. Trends Biochem. Sci. 37, 526–533 (2012).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Chen, F., Mackey, A. J., Stoeckert, C. J. & Roos, D. S. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 34, D363–D368 (2006).
Maric, D. et al. Molecular determinants of ciliary membrane localization of Trypanosoma cruzi flagellar calcium-binding protein. J. Biol. Chem. 286, 33109–33117 (2011).
Saborio, J. L. et al. Isolation and characterization of paraflagellar proteins from Trypanosoma cruzi. J. Biol. Chem. 264, 4071–4075 (1989).
Fouts, D. L. et al. Evidence for Four Distinct Major Protein Components in the Paraflagellar Rod of Trypanosoma cruzi. J. Biol. Chem. 273, 21846–21855 (1998).
Lacomble, S., Portman, N. & Gull, K. A protein-protein interaction map of the Trypanosoma brucei paraflagellar rod. PLoS One 4, e7685 (2009).
Szempruch, A. J. et al. Extracellular Vesicles from Trypanosoma brucei Mediate Virulence Factor Transfer and Cause Host Anemia. Cell 164, 246–257 (2016).
Banks, K. L. Binding of Trypanosoma congolense to the Walls of Small Blood Vessels*. J. Protozool. 25, 241–245 (1978).
Claes, F., Büscher, P., Touratier, L. & Goddeeris, B. M. Trypanosoma equiperdum: master of disguise or historical mistake? Trends Parasitol. 21, 316–321 (2005).
Resh, M. D. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta–Mol. Cell Res. 1451, 1–16 (1999).
Portman, N., Lacomble, S., Thomas, B., McKean, P. G. & Gull, K. Combining RNA interference mutants and comparative proteomics to identify protein components and dependences in a eukaryotic flagellum. J. Biol. Chem. 284, 5610–5619 (2009).
Boman, A. & Kahn, R. Arf proteins: the membrane traffic police? Trends Biochem. Sci. 20, 147–150 (1995).
Amor, J. C., Harrison, D. H., Kahn, R. A. & Ringe, D. Structure of the human ADP-ribosylation factor 1 complexed with GDP. Nature 372, 704–708 (1994).
Tetaud, E. et al. TbFlabarin, a flagellar protein of Trypanosoma brucei, highlights differences between Leishmania and Trypanosoma flagellar-targeting signals. Exp. Parasitol. 166, 97–107 (2016).
Acknowledgements
We thank Elisabeth Kremmer and Keith Gull for providing antibodies and Nicolai Siegel for helpful discussion and critical reading of the manuscript. We thank Margarida Vaz and Fabien Guegan for assistance with the mouse experiments and transfections.
Author information
Authors and Affiliations
Contributions
Z.D., N.E. and S.H. performed the experiments and prepared the Figures 3–7, 9 and 10. M.D. and F.B. performed and analyzed the mass spectrometry experiments. T.S. contributed the phylogenetic analysis. B.M. contributed the cell fractionation experiment. L.M.F., F.B. and C.J.J. wrote the manuscript and supervised the study.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Cicova, Z., Dejung, M., Skalicky, T. et al. Two flagellar BAR domain proteins in Trypanosoma brucei with stage-specific regulation. Sci Rep 6, 35826 (2016). https://doi.org/10.1038/srep35826
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35826
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
