
Abstract
Aims/hypothesis
Epidemiological studies have generated conflicting findings on the relationship between glucose-lowering medication use and cancer risk. Naturally occurring variation in genes encoding glucose-lowering drug targets can be used to investigate the effect of their pharmacological perturbation on cancer risk.
Methods
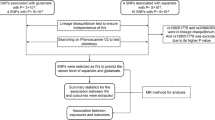
We developed genetic instruments for three glucose-lowering drug targets (peroxisome proliferator activated receptor γ [PPARG]; sulfonylurea receptor 1 [ATP binding cassette subfamily C member 8 (ABCC8)]; glucagon-like peptide 1 receptor [GLP1R]) using summary genetic association data from a genome-wide association study of type 2 diabetes in 148,726 cases and 965,732 controls in the Million Veteran Program. Genetic instruments were constructed using cis-acting genome-wide significant (p<5×10−8) SNPs permitted to be in weak linkage disequilibrium (r2<0.20). Summary genetic association estimates for these SNPs were obtained from genome-wide association study (GWAS) consortia for the following cancers: breast (122,977 cases, 105,974 controls); colorectal (58,221 cases, 67,694 controls); prostate (79,148 cases, 61,106 controls); and overall (i.e. site-combined) cancer (27,483 cases, 372,016 controls). Inverse-variance weighted random-effects models adjusting for linkage disequilibrium were employed to estimate causal associations between genetically proxied drug target perturbation and cancer risk. Co-localisation analysis was employed to examine robustness of findings to violations of Mendelian randomisation (MR) assumptions. A Bonferroni correction was employed as a heuristic to define associations from MR analyses as ‘strong’ and ‘weak’ evidence.
Results
In MR analysis, genetically proxied PPARG perturbation was weakly associated with higher risk of prostate cancer (for PPARG perturbation equivalent to a 1 unit decrease in inverse rank normal transformed HbA1c: OR 1.75 [95% CI 1.07, 2.85], p=0.02). In histological subtype-stratified analyses, genetically proxied PPARG perturbation was weakly associated with lower risk of oestrogen receptor-positive breast cancer (OR 0.57 [95% CI 0.38, 0.85], p=6.45×10−3). In co-localisation analysis, however, there was little evidence of shared causal variants for type 2 diabetes liability and cancer endpoints in the PPARG locus, although these analyses were likely underpowered. There was little evidence to support associations between genetically proxied PPARG perturbation and colorectal or overall cancer risk or between genetically proxied ABCC8 or GLP1R perturbation with risk across cancer endpoints.
Conclusions/interpretation
Our drug target MR analyses did not find consistent evidence to support an association of genetically proxied PPARG, ABCC8 or GLP1R perturbation with breast, colorectal, prostate or overall cancer risk. Further evaluation of these drug targets using alternative molecular epidemiological approaches may help to further corroborate the findings presented in this analysis.
Data availability
Summary genetic association data for select cancer endpoints were obtained from the public domain: breast cancer (https://bcac.ccge.medschl.cam.ac.uk/bcacdata/); and overall prostate cancer (http://practical.icr.ac.uk/blog/). Summary genetic association data for colorectal cancer can be accessed by contacting GECCO (kafdem at fredhutch.org). Summary genetic association data on advanced prostate cancer can be accessed by contacting PRACTICAL (practical at icr.ac.uk). Summary genetic association data on type 2 diabetes from Vujkovic et al (Nat Genet, 2020) can be accessed through dbGAP under accession number phs001672.v3.p1 (pha004945.1 refers to the European-specific summary statistics). UK Biobank data can be accessed by registering with UK Biobank and completing the registration form in the Access Management System (AMS) (https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access).
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.

Introduction
Globally, an estimated 460 million individuals have type 2 diabetes, most of whom require long-term use of glucose-lowering medications to maintain glycaemic control [1]. Several different classes of oral glucose-lowering medications are used to manage this condition, including biguanides (e.g. metformin), sulfonylureas, thiazolidinediones, dipeptidyl peptidase-4 (DPP-4) inhibitors, sodium–glucose cotransporter 2 (SGLT2) inhibitors and glucagon-like peptide-1 receptor (GLP1R) agonists, with diverse mechanisms of action [2].
Preclinical studies have variably reported both carcinogenic and antineoplastic effects of glucose-lowering medications. For example, in vitro studies have suggested that metformin, an insulin sensitiser and first-line therapy for type 2 diabetes, can reduce cell proliferation, induce apoptosis and cause cell cycle arrest [3]. Thiazolidinediones, insulin sensitisers and selective peroxisome proliferator activated nuclear receptor (PPARG) agonists have been suggested to increase cellular differentiation, reduce cellular proliferation and induce apoptosis in some cell lines but to promote metastatic prostate cancer in vivo [4,5,6]. There is also some evidence that sulfonylureas, secretagogues that lower blood glucose levels by stimulating pancreatic insulin secretion, may promote carcinogenesis, potentially via increasing circulating insulin levels [7, 8]. Finally, in vitro studies have reported potential antiproliferative effects of GLP1R agonists in various cancer cell types [9,10,11].
Epidemiological studies of glucose-lowering medication use have provided some support for findings from laboratory studies. For example, some observational studies have reported that metformin users have lower risk of several cancers while sulfonylurea use has been associated with an increased risk of site-specific (i.e. colorectal, metastatic prostate) and overall cancer [12,13,14,15,16,17]. In addition, some thiazolidinediones (i.e. pioglitazones) have been linked to an elevated risk of bladder, prostate and pancreatic cancer, though use of rosiglitazone has been associated with lower breast cancer risk [18, 19]. Finally, GLP1R agonist use has been associated with a decreased risk of prostate cancer when compared with sulfonylurea use [20].
The causal nature of associations reported between glucose-lowering medication use and cancer risk in conventional epidemiological studies is often unclear. This is because of the susceptibility of such studies to residual confounding (e.g. due to indication) and various forms of bias (e.g. immortal time, prevalent user), which can undermine robust causal inference [21]. While clinical trials of glucose-lowering medications have not consistently reported differences in rates of cancer among users of these medications, such studies are often underpowered to detect effects for individual cancer sites [22, 23]. Further, such studies often have limited follow-up periods, thus are not able to adequately capture outcomes with long induction periods, such as cancer.
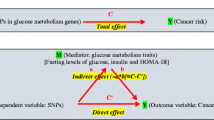
Drug target Mendelian randomisation (MR) uses germline variants in genes encoding drug targets as instruments (‘proxies’) for these targets to estimate the effect of their pharmacological perturbation on disease endpoints [24]. Since germline genetic variants are randomly assorted at meiosis and fixed at conception, analyses using variants as instruments should be less prone to conventional issues of confounding and reverse causation. In addition, given the length of time required for solid tumour development, the use of germline genetic variants as instruments is advantageous as it permits estimation of the long-term effects of medication use on cancer risk [25].
Given the widespread use of glucose-lowering medications and reports of both adverse and protective associations of these medications with cancer risk in preclinical and epidemiological studies, there is a need to further evaluate the role of these medications in the risk of common adulthood cancers. Additionally, given the long induction period of cancers, using MR to examine target-mediated effects of medications that have been on the market for relatively short periods of time (e.g. SGLT2 inhibitors and GLP1R agonists) can be informative in predicting their long-term safety profiles. We thus aimed to develop genetic instruments for the targets of five approved type 2 diabetes medications with known mechanisms of action (sulfonylurea receptor 1 [ATP binding cassette subfamily C member 8 (ABCC8)], PPARG, SGLT2, DPP4 and GLP1R). We also aimed to evaluate associations of genetically proxied perturbation of three of these targets with reliable cis-acting instruments (ABCC8, PPARG and GLP1R) with risk of breast, colorectal and prostate cancer, common cancers with epidemiological evidence suggesting a link between glucose-lowering medication use and their onset, and overall (i.e. site-combined) cancer [5, 12,13,14, 18, 19, 26,27,28].
Methods
Summary genetic association data were obtained from three cancer-specific genome-wide association study (GWAS) consortia. Summary genetic association estimates for overall and oestrogen receptor (ER)-stratified breast cancer risk in up to 122,977 cases and 105,974 controls were obtained from the Breast Cancer Association Consortium (BCAC) [29]. Summary genetic association estimates for overall and site-specific (i.e. colon, rectal) colorectal cancer risk in up to 58,221 cases and 67,694 controls were obtained from an analysis of the Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO), Colorectal Transdisciplinary Study (CORECT), and Colon Cancer Family Registry (CCFR) [30]. Summary genetic association estimates for overall and advanced prostate cancer risk (i.e. metastatic disease, Gleason score ≥8, prostate-specific antigen >100 or prostate cancer-related death) in up to 79,148 cases and 61,106 controls were obtained from the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) consortium [31]. These analyses were restricted to participants of European ancestry.
Overall (i.e. site-combined) cancer risk data in 27,483 incident cases and 372,016 controls were also obtained from a GWAS performed in the UK Biobank cohort study [32]. Briefly, cancer cases were classified according to ICD-9 (http://www.icd9data.com/2007/Volume1/default.htm) and ICD-10 (http://apps.who.int/classifications/icd10/browse/2016/en) with data completed to April 2019 and controls were defined as individuals who did not have any cancer code (ICD9 or ICD10) and did not self-report a cancer diagnosis. GWAS were performed using a linear mixed model as implemented in BOLT-LMM (v2.3) (to account for relatedness and population stratification) and adjusted for age, sex and genotyping array [33]. Further information on imputation and quality control measures have been reported elsewhere [33].
Further information on statistical analysis, imputation, and quality control measures for summary genetic association data obtained from cancer consortia is available in the original publications. All studies contributing data to these analyses had the relevant institutional review board approval from each country, in accordance with the Declaration of Helsinki, and all participants provided informed consent.
Instrument construction
To generate genetic instruments to proxy glucose-lowering drug target perturbation, summary genetic association data were obtained from a GWAS of type 2 diabetes in the Million Veteran Program (148,726 cases; 965,732 controls of European ancestry) [34]. Analyses were adjusted for age, sex and ten principal components of genetic ancestry. Instruments were constructed in PLINK by obtaining SNPs associated with type 2 diabetes at genome-wide significance (p<5×10−8) that were in or within ±500 kb from the gene encoding each respective target (PPARG, Chr3: 12328867–12475855; ABCC8, Chr11: 17414432–17498449; GLP1R, Chr6: 39016574–39055519) using the 1000 Genomes Phase 3 reference panel [35, 36]. We were unable to identify genome-wide significant SNPs within 500 kb windows from SLC5A2 and DPP4 (i.e. instruments for SGLT2 and DPP-4 inhibitors, respectively) and therefore did not proceed with MR analyses for these targets. We also did not include putative metformin targets due to the unclear mechanism(s) of action of this medication [37]. For PPARG, ABCC8 and GLP1R, SNPs used as instruments were permitted to be in weak linkage disequilibrium (r2<0.20) with each other to increase the proportion of variance in each respective drug target explained by the instrument, maximising instrument strength. In total, nine SNPs that met these criteria were obtained for PPARG, six for ABCC8 and four for GLP1R.
In a separate population (i.e. the UK Biobank cohort study), we then evaluated the association of type 2 diabetes SNPs in drug target regions with HbA1c levels, a marker of long-term blood glucose levels, in order to minimise winner’s curse bias. The UK Biobank is a prospective cohort study of ~500,000 individuals aged 40–69 years when recruited in 2006–2010 [38]. SNP summary statistics were re-scaled to represent a mmol/mol (0.09%) unit reduction in HbA1c to provide more interpretable effect estimates in MR analyses. HbA1c values were obtained from a GWAS of 407,766 participants of the UK Biobank performed using a linear mixed model as implemented in BOLT-LMM and adjusted for age, sex, batch and ten principal components of genetic ancestry. For the purposes of this analysis, we sequentially removed participants according to the following exclusion criteria: withdrawn from the study (N=502,506 retained); non-European ancestry (N=462,898 retained); missing HbA1c data (N=442,529 retained); missing or ‘prefer not to answer’ response to self-reported diabetes status (N=442,268); self-reported diabetes diagnosis (N=418,574); ICD-10 diabetes diagnosis (N=409,812); missing data on glucose-lowering medication use (N=409,762); self-reported glucose-lowering medication use (N=409,614); HbA1c >48 mmol/mol (6.5%) (N=408,319); and HbA1c <21.88 mmol/mol (4.2%) (N=407,766). Further information on imputation and quality control measures have been reported elsewhere [39].
For the PPARG instrument, two SNPs where the effect on HbA1c was in the opposite direction to that of type 2 diabetes were removed from the instrument (rs17036160, rs11712085), as these associations likely represent pleiotropic mechanisms that would bias consequent MR analyses.
Instrument validation
Instruments were validated by examining the association of genetically proxied drug target perturbation with endpoints influenced by these medications in randomised controlled trials. For PPARG, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were used as positive controls (i.e. PPARG agonists lower levels of ALT and AST) and for ABCC8 and GLP1R, BMI was used (i.e. sulfonylureas cause weight gain and GLP1R agonists cause weight loss) [40,41,42,43]. Co-localisation was then performed to assess whether genetic liability to type 2 diabetes and traits representing positive controls share the same causal variant at each locus encoding a drug target (i.e. PPARG, ABCC8, GLP1R). Such an analysis can permit exploration of whether genetic liability to type 2 diabetes and positive control traits at each drug target locus are influenced by distinct causal variants that are in linkage disequilibrium with each other, indicative of horizontal pleiotropy (an instrument influencing an outcome through pathways independent to that of the exposure), a violation of the exclusion restriction criterion.
Co-localisation analysis was performed using the coloc (version 2.0) R package (https://cran.r-project.org/web/packages/coloc/index.html), which uses approximate Bayes factor computation to generate posterior probabilities that associations between two traits represent each of the following configurations: (1) neither trait has a genetic association in the region (H0); (2) only the first trait has a genetic association in the region (H1); (3) only the second trait has a genetic association in the region (H2); (4) both traits are associated but have different causal variants (H3); and (5) both traits are associated and share a single causal variant (H4) [44]. Co-localisation analysis was performed by generating ±500 kb windows around the gene encoding each respective drug target. We used a posterior probability of >50% to indicate support for a configuration tested. Where there was not support for H4, we then examined the possibility of co-localisation across other secondary conditionally independent signals for either genetic liability to type 2 diabetes or positive controls within drug target loci by performing pairwise conditional and co-localisation analysis on all conditionally independent association signals using GCTA-COJO and the coloc package as implemented in pwCoCo [45]. We employed default priors for p1 (i.e. prior probability that a SNP is associated with type 2 diabetes liability within a drug target locus, 1×10−4), p2 (i.e. prior probability that a SNP is associated with positive controls or cancer risk within a drug target locus, 1×10−4) and p12 (i.e. prior probability that a SNP is associated with both traits, 1×10−5). As sensitivity analyses, we re-performed co-localisation analysis employing two alternate priors for p12 (5×10−5, 5×10−6).
Statistical analysis
Causal estimates were generated using inverse-variance weighted (IVW) random-effects models (permitting overdispersion in models). These models were adjusted for weak linkage disequilibrium between SNPs with reference to the 1000 Genomes Phase 3 reference panel [46]. Where there was under-dispersion in causal estimates generated from individual genetic variants, the residual SE was set to 1 (i.e. equivalent to a fixed-effects model).
MR analysis makes the following assumptions: (1) that a genetic instrument is associated with a modifiable exposure or drug target (‘relevance’); (2) the instrument does not share a common cause with an outcome (‘exchangeability’); and (3) the instrument has no direct effect on the outcome (‘exclusion restriction’).
The ‘relevance’ MR assumption was evaluated by generating estimates of the proportion of variance of each drug target (in HbA1c units) explained by the instrument (r2) and F statistics. F statistics can be used to examine whether results are likely to be influenced by weak instrument bias (i.e. reduced statistical power and bias when an instrument explains a limited proportion of the variance in a drug target). As a convention, an F statistic of >10 is indicative of minimal weak instrument bias.
We evaluated the ‘exclusion restriction’ MR assumption by performing co-localisation to examine whether drug targets and cancer endpoints showing nominal evidence of an association in MR analyses (p<0.05) share the same causal variant at a given locus. Iterative leave-one-out analysis was performed by removing one SNP at a time from instruments to examine whether findings showing nominal evidence of association were driven by a single influential SNP.
To account for multiple testing across analyses, a Bonferroni correction was used to establish a p value threshold of <0.0019 (false-positive rate = 0.05/27 statistical tests [three drug targets tested against nine primary cancer endpoints]), which we used as a heuristic to define ‘strong evidence’, with findings between p≥0.0019 and p<0.05 defined as ‘weak evidence’.
There was no formal prespecified protocol for this study. All statistical analyses were performed using R version 3.3.1 (https://www.r-project.org/).
Results
Characteristics of genetic variants used to instrument glucose-lowering drug targets are presented in Table 1. Across all three drug targets, F statistics for their respective instruments ranged from 56.32 to 487.14, suggesting that weak instrument bias was unlikely to affect the conclusions (ESM Table 1). Power calculations suggested that we had 80% power to detect ORs ranging from 1.40 to 2.62 (in PPARG analyses), 2.03 to 8.34 (in ABCC8 analyses) and 2.22 to 8.78 (in GLP1R analyses) per mmol/mol reduction in target-mediated inverse rank normal transformed [IRNT] HbA1c across all cancer endpoints (α=0.05). Complete power estimates across all MR analyses are presented in ESM Table 2.
Instrument validation
Genetically proxied PPARG perturbation was associated with lower levels of ALT (SD change in ALT per PPARG perturbation equivalent to 1 unit IRNT HbA1c reduction: −0.57 [95% CI −1.01, −0.13], p=0.01) and AST (−0.49 [95% CI −1.79, −0.19], p=1.53×10−3). Co-localisation analysis suggested that type 2 diabetes associations in the PPARG locus had a 92% and 84% probability of sharing a causal variant with ALT and AST, respectively (Figs 1, 2 and 3 and ESM Tables 3, 4).

Regional Manhattan plot of associations of SNPs with type 2 diabetes ±500 kb from the PPARG locus. rs17036160 (purple dot) represents the sentinel SNP associated with genetic liability to type 2 diabetes in the PPARG locus

Regional Manhattan plot of associations of SNPs with ALT concentrations ±500 kb from the PPARG locus. rs17036160 (purple dot) represents the sentinel SNP associated with genetic liability to type 2 diabetes in the PPARG locus. SNPs in unclear linkage disequilibrium with sentinel SNP are in grey

Regional Manhattan plot of associations of SNPs with AST concentrations ±500 kb from the PPARG locus. rs17036160 (purple dot) represents the sentinel SNP associated with genetic liability to type 2 diabetes in the PPARG locus
Genetically proxied ABCC8 perturbation was associated with elevated BMI (SD change in BMI per ABCC8 perturbation equivalent to 1 unit IRNT HbA1c reduction: 0.530 [95% CI 0.004, 0.172], p=3.75×10−3). Co-localisation analysis suggested that type 2 diabetes and BMI associations had a 94.0% posterior probability of sharing a causal variant in ABCC8 (Figs 4, 5 and ESM Table 5).

Regional Manhattan plot of associations of SNPs with type 2 diabetes ±500 kb from the ABCC8 locus. rs5219 (purple dot) represents the sentinel SNP associated with genetic liability to type 2 diabetes in the ABCC8 locus

Regional Manhattan plot of associations of SNPs with BMI ±500 kb from the ABCC8 locus. rs5219 (purple dot) represents the sentinel SNP associated with genetic liability to type 2 diabetes in the ABCC8 locus
There was little evidence to support an association of genetically proxied GLP1R perturbation with BMI (SD change in BMI equivalent to 1 unit IRNT HbA1c reduction: −0.08 [95% CI −0.30, 0.15], p=0.51). Co-localisation analysis applied to both marginal and conditionally independent associations for type 2 diabetes and BMI in the GLP1R locus did not support shared causal variants across these traits (posterior probability of shared causal variants across models: 0.22–0.49%) (Figs 6, 7 and ESM Table 6).

Regional Manhattan plot of associations of SNPs with type 2 diabetes ±500 kb from the GLP1R locus. rs10305420 (purple dot) represents the sentinel SNP associated with genetic liability to type 2 diabetes in the GLP1R locus

Regional Manhattan plot of associations of SNPs with BMI ±500 kb from the GLP1R locus. rs10305420 (purple dot) represents the sentinel SNP associated with genetic liability to type 2 diabetes in the GLP1R locus
Genetically proxied PPARG perturbation and cancer risk
There was weak evidence for an association of genetically proxied PPARG perturbation with an elevated risk of prostate cancer (OR 1.75 [95% CI 1.07, 2.85], p=0.02) but little evidence of association with other cancer endpoints (Table 2). Findings for prostate cancer risk were consistent in iterative leave-one-out analysis (ESM Table 7). Co-localisation using marginal and conditional associations for type 2 diabetes and prostate cancer in the PPARG locus suggested that type 2 diabetes was unlikely to share a causal variant with this cancer in this region (posterior probability of a shared causal variant across models: ≤0.09%, posterior probability of distinct causal variants: ≤25%) (Fig. 8 and ESM Table 8).

Regional Manhattan plot of associations of SNPs with prostate cancer risk ±500 kb from the PPARG locus. rs17036160 (purple dot) represents the sentinel SNP associated with genetic liability to type 2 diabetes in the PPARG locus
In subtype-stratified analyses, genetically proxied PPARG perturbation was weakly associated with lower risk of ER+ breast cancer (OR 0.57 [95% CI 0.38, 0.85], p=6.45×10−3). This finding was consistent in iterative leave-one-out analysis (ESM Table 9). Co-localisation using marginal and conditional associations for type 2 diabetes and ER+ breast cancer in the PPARG locus reported a low posterior probability (H4<5%; posterior probability of distinct causal variants: ≤23%) of both traits sharing one or more causal variants within this region (Fig. 9 and ESM Table 10).

Regional Manhattan plot of associations of SNPs with ER+ breast cancer risk ±500 kb from the PPARG locus. rs17036160 (purple dot) represents the sentinel SNP associated with genetic liability to type 2 diabetes in the PPARG locus
Genetically proxied ABCC8 and GLP1R perturbation and cancer risk
There was little MR evidence of association of genetically proxied ABCC8 or GLP1R perturbation with site-specific or overall cancer risk (Tables 3, 4).
Sensitivity analyses altering priors for co-localisation
Across positive control traits and cancer outcomes, findings from co-localisation analyses remained robust to using two alternate priors for p12 (5×10−5, 5×10−6) (ESM Table 11).
Discussion
In this MR analysis of up to 287,829 cases and 606,790 controls, we found weak evidence for an association of genetically proxied PPARG perturbation with a higher risk of prostate cancer and lower risk of ER+ breast cancer. In co-localisation analysis, however, there was little evidence that genetic liability to type 2 diabetes and these cancer endpoints shared one or more causal variants within PPARG, though these analyses were likely underpowered given low posterior probabilities to support both H3 (i.e. distinct causal variants) and H4 (i.e. shared causal variants) across these analyses. We found little evidence of association of genetically proxied GLP1R or ABCC8 perturbation with cancer risk.
Despite in vivo studies suggesting an important role for PPARG in prostate tumour growth and conventional epidemiological studies suggesting a link between pioglitazone use and elevated prostate cancer risk, our combined MR and co-localisation analyses did not find consistent evidence for an association of genetically proxied PPARG perturbation with prostate cancer risk [6, 18]. Likewise, our findings are not consistent with some previous epidemiological studies that have reported links between rosiglitazone use and lower breast cancer risk and thiazolidinedione use and lower colorectal cancer risk [5, 19]. Though our analyses were powered to detect effect sizes comparable with those reported in some previous studies (e.g. ~60% increased prostate cancer risk among pioglitazone users and ~60% lower risk of colorectal cancer among thiazolidinedione users), they were likely less powered to detect other, more modest, effect sizes reported in the literature (e.g. ~10% lower risk of breast cancer in rosiglitazone users) [19, 26, 47]. Interpretation of the pharmacoepidemiological literature linking glucose-lowering medication use with cancer risk is challenging because of the likely susceptibility of many previous studies to residual confounding (e.g. by indication) due to the use of inappropriate comparator groups (i.e. non-medication users), the inclusion of ‘prevalent users’ of medications in analyses and the possibility of ‘immortal time’ bias arising due to misalignment of the start of follow-up, eligibility and treatment assignment of participants [21].
Among the strengths of our analysis is the strict instrument selection and validation process employed. By using cis-acting variants, in close proximity to the genes that code for the drug targets of interest, horizontal pleiotropy should be minimised. In addition, we used strict positive control analysis (i.e. testing drug targets against established secondary effects of medications) and co-localisation analyses (including co-localisation analyses permitting multiple causal variants) to validate the selected instruments. Our use of a summary-data MR approach permitted us to leverage large-scale genetic data from several GWAS consortia, enhancing statistical power and precision of causal estimates.
There were several limitations to this analysis. First, we had sufficient statistical power to detect large effect sizes only per SD decrease in HbA1c (~6.75 mmol/mol [~0.61%]) and therefore cannot rule out more modest effects of the drug targets examined on cancer risk. In clinical trials, monotherapy with sulfonylureas, thiazolidinediones (rosiglitazone, pioglitazone) and the GLP1R agonist liraglutide has been shown to reduce HbA1c by around 8–17 mmol/mol (0.7–1.5%), as compared with placebo [48,49,50]. Second, although co-localisation analyses of PPARG and cancer endpoints provided low posterior probabilities for shared causal variants, it should be noted that this may also reflect limited power. The low posterior probabilities supporting either shared or distinct causal variants across several co-localisation analyses suggests that many of these analyses may have been too underpowered to support either of these configurations evaluated. Third, the low posterior probability of shared causal variants in ‘positive control’ co-localisation analyses for GLP1R and BMI could reflect distinct signalling mechanisms influencing type 2 diabetes and BMI in GLP1R, the presence of which would not necessarily influence the validity of this as an instrument for GLP1R signalling perturbation’s effect on glycaemic control [51]. Fourth, we were unable to evaluate the role of some glucose-lowering drug targets (i.e. DPP-4 and SGLT2) due to the absence of reliable genetic instruments for these targets. Fifth, our analyses were restricted to the examination of target-mediated (i.e. ‘on-target’) effects of glucose-lowering medications on cancer endpoints. Sixth, our analyses assume no gene–environment or gene–gene interactions and linear and time-dependent effects of drug targets on cancer risk. Seventh, though associations of genetically proxied PPARG perturbation and prostate and ER+ breast cancer risk attenuated towards the null in iterative leave-one-out analysis removing rs4135247 from the PPARG instrument, 95% CIs overlapped across models with and without this variant. Though this attenuation in association is consistent with sampling error, we cannot rule out the possibility that this attenuation was driven, in part, through horizontally pleiotropic mechanisms linking this variant to cancer risk. Eighth, though we found strong and suggestive evidence for associations of genetically proxied PPARG perturbation with ER+ breast cancer and prostate cancer risk, respectively, after applying a Bonferroni correction to account for multiple testing, we cannot rule out the possibility that these findings represent false-positive results. Ninth, the MR estimates reported represent long-term effects of target modulation in non-diabetic populations, whereas the clinical effects of these medications may be more pronounced among individuals with type 2 diabetes and could depend on length of medication use. Tenth, we cannot rule out the possibility that controls in cancer GWAS included individuals with latent, undiagnosed cancer, the presence of which would bias associations towards or away from the null, depending on the site of undiagnosed cancer and the relationship between drug targets examined and this cancer. We also cannot rule out the possibility of survival bias influencing genetic association estimates from cancer GWAS consortia that employed case–control study designs. If, for example, genetic variants used to instrument glucose-lowering drug target perturbation increased cancer risk and subsequent mortality prior to enrolment in a case–control study, this could induce an artificial ‘protective’ association between perturbation of this drug target and cancer risk. Finally, samples were restricted to individuals of European ancestry and therefore the generalisability of these findings to non-European populations is unclear.
In conclusion, we developed novel instruments for PPARG, ABCC8 and GLP1R using strict validation protocols and evaluated the association of genetically proxied perturbation of these targets with risk of cancer. In MR analysis we found weak evidence that genetically proxied PPARG perturbation was associated with a higher risk of prostate cancer and a lower risk of ER+ breast cancer. There was little evidence of co-localisation for these findings, a necessary precondition to infer causality between PPARG perturbation and these cancer endpoints, possibly reflecting either the absence of shared causal variants across type 2 diabetes liability and these cancer endpoints in PPARG or the low statistical power of these analyses. Further assessment of these drug targets using alternative molecular epidemiological approaches (e.g. using protein or expression quantitative trait loci or using direct circulating measures of these proteins) and/or studies using medical registry data (e.g. ‘target trial’ analyses) may help to further corroborate findings presented in this analysis. Finally, we found little evidence for an association of genetically proxied ABCC8 and GLP1R perturbation with risk of breast, colorectal, prostate or overall cancer risk.
Abbreviations
- ABCC8:
-
Sulfonylurea receptor 1 (ATP binding cassette subfamily C member 8)
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- BCAC:
-
Breast Cancer Association Consortium
- CCFR:
-
Colon Cancer Family Registry
- CORECT:
-
Colorectal Transdisciplinary Study
- DPP-4:
-
Dipeptidyl peptidase-4
- ER:
-
Oestrogen receptor
- GECCO:
-
Genetics and Epidemiology of Colorectal Cancer Consortium
- GLP1R:
-
Glucagon-like peptide-1 receptor
- GWAS:
-
Genome-wide association study
- IRNT:
-
Inverse rank normal transformed
- MR:
-
Mendelian randomisation
- PPARG:
-
Peroxisome proliferator activated nuclear receptor γ
- PRACTICAL:
-
Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome
- SGLT2:
-
Sodium–glucose cotransporter 2
References
Khan MAB, Hashim MJ, King JK, Govender RD, Mustafa H, Al Kaabi J (2020) Epidemiology of type 2 diabetes - global burden of disease and forecasted trends. J Epidemiol Glob Health 10(1):107–111
Chaudhury A, Duvoor C, Reddy Dendi VS et al (2017) Clinical review of antidiabetic drugs: implications for type 2 diabetes mellitus management. Front Endocrinol (Lausanne) 8:6
Lei Y, Yi Y, Liu Y et al (2017) Metformin targets multiple signaling pathways in cancer. Chin J Cancer 36(1):17. https://doi.org/10.1186/s40880-017-0184-9
Chou FS, Wang P-S, Kulp S, Pinzone JJ (2007) Effects of thiazolidinediones on differentiation, proliferation, and apoptosis. Mol Cancer Res 5(6):523–30. https://doi.org/10.1158/1541-7786.MCR-06-0278
Liu Y, Jin P-P, Sun X-C, Hu T-T (2018) Thiazolidinediones and risk of colorectal cancer in patients with diabetes mellitus: a meta-analysis. Saudi J Gastroenterol 24(2):75–81. https://doi.org/10.4103/sjg.SJG_295_17
Ahmad I, Mui E, Galbraith L et al (2016) Sleeping Beauty screen reveals Pparg activation in metastatic prostate cancer. Proc Natl Acad Sci U S A 113(29):8290–5. https://doi.org/10.1073/pnas.1601571113
Ferguson RD, Novosyadlyy R, Fierz Y et al (2012) Hyperinsulinemia enhances c-Myc-mediated mammary tumor development and advances metastatic progression to the lung in a mouse model of type 2 diabetes. Breast Cancer Res 14(1):R8. https://doi.org/10.1186/bcr3089
Wu X, Fan Z, Masui H, Rosen N, Mendelsohn J (1995) Apoptosis induced by an anti-epidermal growth factor receptor monoclonal antibody in a human colorectal carcinoma cell line and its delay by insulin. J Clin Invest 95(4):1897–905. https://doi.org/10.1172/JCI117871
Koehler JA, Kain T, Drucker DJ (2011) Glucagon-like peptide-1 receptor activation inhibits growth and augments apoptosis in murine CT26 colon cancer cells. Endocrinology 152(9):3362–72. https://doi.org/10.1210/en.2011-1201
Ligumsky H, Wolf I, Israeli S et al (2012) The peptide-hormone glucagon-like peptide-1 activates cAMP and inhibits growth of breast cancer cells. Breast Cancer Res Treat 132(2):449–61. https://doi.org/10.1007/s10549-011-1585-0
Nomiyama T, Kawanami T, Irie S et al (2014) Exendin-4, a GLP-1 receptor agonist, attenuates prostate cancer growth. Diabetes 63(11):3891–905. https://doi.org/10.2337/db13-1169
Haring A, Murtola TJ, Talala K, Taari K, Tammela TLJ, Auvinen A (2017) Antidiabetic drug use and prostate cancer risk in the Finnish Randomized Study of Screening for Prostate Cancer. Scand J Urol 51(1):5–12. https://doi.org/10.1080/21681805.2016.1271353
Shin CM, Kim N, Han K (2020) Anti-diabetic medications and the risk for colorectal cancer: A population-based nested case-control study. Cancer Epidemiol 64:101658
Wu L, Zhu J, Prokop LJ, Murad MH (2015) Pharmacologic therapy of diabetes and overall cancer risk and mortality: a meta-analysis of 265 studies. Sci Rep 5:10147. https://doi.org/10.1038/srep10147
Decensi A, Puntoni M, Goodwin P et al (2010) Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) 3(11):1451–61. https://doi.org/10.1158/1940-6207.CAPR-10-0157
Noto H, Goto A, Tsujimoto T, Noda M (2012) Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One 7(3):e33411
Zhang K, Bai P, Dai H, Deng Z (2021) Metformin and risk of cancer among patients with type 2 diabetes mellitus: a systematic review and meta-analysis. Prim Care Diabetes 15(1):52–58. https://doi.org/10.1016/j.pcd.2020.06.001
Qu H, Zheng Y, Wang Y (2017) Global and regional effects of bladder cancer risk associated with pioglitazone therapy in patients with diabetes. Scientific Rep 7(1):15804–15804. https://doi.org/10.1038/s41598-017-16074-1
Tseng CH (2017) Rosiglitazone reduces breast cancer risk in Taiwanese female patients with type 2 diabetes mellitus. Oncotarget 8(2):3042–3048. https://doi.org/10.18632/oncotarget.13824
Lu S, Yin H, Yu OHY, Azoulay L (2022) Incretin-based drugs and the incidence of prostate cancer among patients with type 2 diabetes. Epidemiology 33(4):563–571. https://doi.org/10.1097/EDE.0000000000001486
McMahon AD (2003) Approaches to combat with confounding by indication in observational studies of intended drug effects. Pharmacoepidemiol Drug Saf 12(7):551–558. https://doi.org/10.1002/pds.883
Dicembrini I, Nreu B, Mannucci E, Monami M (2019) Sodium-glucose co-transporter-2 (SGLT-2) inhibitors and cancer: A meta-analysis of randomized controlled trials. Diabetes Obes Metab 21(8):1871–1877. https://doi.org/10.1111/dom.13745
Piccoli GF, Mesquita LA, Stein C et al (2020) Do GLP-1 receptor agonists increase the risk of breast cancer? A systematic review and meta-analysis. J Clin Endocrinol Metab 106(3):912–921
Yarmolinsky J, Wade KH, Richmond RC et al (2018) causal inference in cancer epidemiology: what is the role of Mendelian randomization? Cancer Epidemiol Biomarkers Prev 27(9):995–1010. https://doi.org/10.1158/1055-9965.EPI-17-1177
Nadler DL, Zurbenko IG (2013) Developing a Weibull model extension to estimate cancer latency. ISRN Epidemiol 2013:750857
Lin HC, Hsu YT, Kachingwe BH, Hsu CY, Uang YS, Wang LH (2014) Dose effect of thiazolidinedione on cancer risk in type 2 diabetes mellitus patients: a six-year population-based cohort study. J Clin Pharm Ther 39(4):354–360. https://doi.org/10.1111/jcpt.12151
Wang J, Kim CH (2022) Differential risk of cancer associated with glucagon-like peptide-1 receptor agonists: analysis of real-world databases. Endocr Res 47(1):18–25. https://doi.org/10.1080/07435800.2021.1955255
Yang X, So WY, Ma RC et al (2010) Use of sulphonylurea and cancer in type 2 diabetes-The Hong Kong Diabetes Registry. Diabetes Res Clin Pract 90(3):343–351. https://doi.org/10.1016/j.diabres.2010.08.022
Michailidou K, Lindström S, Dennis J et al (2017) Association analysis identifies 65 new breast cancer risk loci. Nature 551(7678):92–94. https://doi.org/10.1038/nature24284
Huyghe JR, Bien SA, Harrison TA et al (2019) Discovery of common and rare genetic risk variants for colorectal cancer. Nat Genet 51(1):76–87. https://doi.org/10.1038/s41588-018-0286-6
Schumacher FR, Al Olama AA, Berndt SI et al (2018) Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet 50(7):928–936. https://doi.org/10.1038/s41588-018-0142-8
Burrows K, Kazmi N, Haycock P, Tsilidis KK, Martin RM, Lewis SJ (2020) Mendelian randomisation study exploring the associations of serum folate with pan and site-specific cancers. bioRxiv 762138
Burrows KB, Haycock P (2021) Genome-wide association study of cancer risk in UK Biobank. University of Bristol. https://doi.org/10.5523/bris.aed0u12w0ede20olb0m77p4b9
Vujkovic M, Keaton JM, Lynch JA et al (2020) Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet 52(7):680–691
Auton A, Brooks LD, Durbin RM et al (2015) A global reference for human genetic variation. Nature 526(7571):68–74. https://doi.org/10.1038/nature15393
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. https://doi.org/10.1186/s13742-015-0047-8
LaMoia TE, Shulman GI (2021) Cellular and molecular mechanisms of metformin action. Endocr Rev 42(1):77–96. https://doi.org/10.1210/endrev/bnaa023
Sudlow C, Gallacher J, Allen N et al (2015) UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12(3):e1001779
Mitchell R, Elsworth BL, Mitchell R et al (2019) MRC IEU UK Biobank GWAS pipeline version 2. University of Bristol. https://doi.org/10.5523/bris.pnoat8cxo0u52p6ynfaekeigi
Vilsbøll T, Christensen M, Junker AE, Knop FK, Gluud LL (2012) Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. BMJ 344:d7771
Phung OJ, Scholle JM, Talwar M, Coleman CI (2010) Effect of noninsulin antidiabetic drugs added to metformin therapy on glycemic control, weight gain, and hypoglycemia in type 2 diabetes. JAMA 303(14):1410–1418. https://doi.org/10.1001/jama.2010.405
Aithal GP, Thomas JA, Kaye PV et al (2008) Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology 135(4):1176–1184. https://doi.org/10.1053/j.gastro.2008.06.047
Ono M, Ikegami H, Fujisawa T et al (2005) Improvement of liver function parameters in patients with type 2 diabetes treated with thiazolidinediones. Metabolism 54(4):529–532. https://doi.org/10.1016/j.metabol.2004.11.007
Giambartolomei C, Vukcevic D, Schadt EE et al (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10(5):e1004383
Zheng J, Haberland V, Baird D et al (2020) Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat Genet 52(10):1122–1131
Burgess S, Dudbridge F, Thompson SG (2016) Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med 35(11):1880–1906. https://doi.org/10.1002/sim.6835
Erdmann E, Harding S, Lam H, Perez A (2016) Ten-year observational follow-up of PROactive: a randomized cardiovascular outcomes trial evaluating pioglitazone in type 2 diabetes. Diabetes Obes Metab 18(3):266–273. https://doi.org/10.1111/dom.12608
Hirst JA, Farmer AJ, Dyar A, Lung TW, Stevens RJ (2013) Estimating the effect of sulfonylurea on HbA1c in diabetes: a systematic review and meta-analysis. Diabetologia 56(5):973–984. https://doi.org/10.1007/s00125-013-2856-6
Lind M, Hirsch IB, Tuomilehto J et al (2015) Liraglutide in people treated for type 2 diabetes with multiple daily insulin injections: randomised clinical trial (MDI Liraglutide trial). BMJ 351:h5364
Mearns ES, Saulsberry WJ, White CM et al (2015) Efficacy and safety of antihyperglycaemic drug regimens added to metformin and sulphonylurea therapy in type 2 diabetes: a network meta-analysis. Diabet Med 32(12):1530–1540. https://doi.org/10.1111/dme.12837
Gill D, Vujkovic M (2022) The potential of genetic data for prioritizing drug repurposing efforts. Neurology 99(7):267–268. https://doi.org/10.1212/WNL.0000000000200803
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Acknowledgements
The authors would like to thank the participants of the individual studies contributing to the BCAC and the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) for their participation in these studies, along with the principal investigators of these consortia for generating the data used for this analysis and for making these data available in the public domain. Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO)-specific acknowledgements include French Association STudy Evaluating RISK for Sporadic Colorectal Cancer (ASTERISK); we are very grateful to B. Buecher without whom this project would not have existed. We also thank all participants in this study, as well as all the physicians, technicians and students. The CCFR graciously thanks the generous contributions of their study participants and acknowledge the dedication of study staff and the financial support from the US National Cancer Institute, without which this important registry would not exist. The authors would like to thank the study participants and staff of the Seattle CCFR and the Hormones and Colon Cancer study (CORE Studies). We thank the participants of Clue II and appreciate the continued efforts of the staff at the Johns Hopkins George W. Comstock Center for Public Health Research and Prevention in the conduct of the Clue II Cohort Study. The authors would like to thank the COLON and NQplus investigators at Wageningen University & Research and the involved clinicians in the participating hospitals. We kindly thank all individuals who agreed to participate in the Colorectal Cancer Study of Austria (CORSA) study. Furthermore, we thank all cooperating physicians and students and the Biobank Graz of the Medical University of Graz. The authors thank the CPS-II participants and Study Management Group for their invaluable contributions to this research. The authors would also like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries, and cancer registries supported by the National Cancer Institute Surveillance Epidemiology and End Results programme. For the Czech Republic Colorectal Cancer Study (CCS), we are thankful to all clinicians in major hospitals in the Czech Republic, without whom the study would not have been practicable. We are also sincerely grateful to all participants in this study. We thank all participants and cooperating clinicians in Darmkrebs: Chancen der Verhütung durch Screening (DACHS), and everyone who provided excellent technical assistance. We acknowledge all contributors to the development of the Early Detection Research Network (EDRN) resource at University of Pittsburgh School of Medicine, Department of Gastroenterology, Department of Pathology, Hepatology and Nutrition and Biomedical Informatics. For European Prospective Investigation into Cancer and Nutrition (EPIC), where authors are identified as personnel of the International Agency for Research on Cancer/WHO, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer/WHO. We are sincerely grateful to all participants who were recruited as part of the EPICOLON project. We acknowledge the Spanish National DNA Bank, Biobank of Hospital Clínic–IDIBAPS and Biobanco Vasco for the availability of the samples. The work was carried out (in part) at the Esther Koplowitz Centre, Barcelona. For the Harvard cohorts (HPFS, NHS, PHS), the study protocol was approved by the institutional review boards of the Brigham and Women’s Hospital and Harvard T. H. Chan School of Public Health, and those of participating registries as required. We acknowledge Channing Division of Network Medicine, Department of Medicine, Brigham and Women’s Hospital as home of the NHS. The authors would like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention’s National Program of Cancer Registries (NPCR) and/or the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) Program. Central registries may also be supported by state agencies, universities and cancer centres. Participating central cancer registries include Alabama, Alaska, Arizona, Arkansas, California, Colorado, Connecticut, Delaware, Florida, Georgia, Hawaii, Idaho, Indiana, Iowa, Kentucky, Louisiana, Massachusetts, Maine, Maryland, Michigan, Mississippi, Montana, Nebraska, Nevada, New Hampshire, New Jersey, New Mexico, New York, North Carolina, North Dakota, Ohio, Oklahoma, Oregon, Pennsylvania, Puerto Rico, Rhode Island, Seattle SEER Registry, South Carolina, Tennessee, Texas, Utah, Virginia, West Virginia and Wyoming. The authors assume full responsibility for analyses and interpretation of data from these registries. We would also like to acknowledge the staff at the Kentucky Cancer Registry. We acknowledge the contributions of Jennifer Barrett, Robin Waxman, Gillian Smith and Emma Northwood in conducting the Leeds Colorectal Cancer Study (LCCS) study. We would like to thank North Carolina Case-Control Study (NCCCS) I and NCCCS II study participants, and the NC Colorectal Cancer Study staff. Northern Swedish Health and Disease Study (NSHDS) investigators thank the Västerbotten Intervention Programme, the Northern Sweden Monitoring of Trends and Determinants in Cardiovascular Disease (MONICA) study, the Biobank Research Unit at Umeå University and Biobanken Norr at Region Västerbotten for providing data and samples and acknowledge the contribution from Biobank Sweden, supported by the Swedish Research Council. The authors thank the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial screening centre investigators and the staff from Information Management Services Inc and Westat Inc. Most importantly, we thank the study participants for their contributions that made this study possible. Cancer incidence data have been provided by the District of Columbia Cancer Registry, Georgia Cancer Registry, Hawaii Cancer Registry, Minnesota Cancer Surveillance System, Missouri Cancer Registry, Nevada Central Cancer Registry, Pennsylvania Cancer Registry, Texas Cancer Registry, Virginia Cancer Registry and Wisconsin Cancer Reporting System. All are supported in part by funds from the Center for Disease Control and Prevention, National Program for Central Registries, local states, or by the National Cancer Institute, Surveillance, Epidemiology and End Results programme. The results reported here and the conclusions derived are the sole responsibility of the authors. We thank the Studies of Epidemiology and Risk Factors in Cancer Heredity (SEARCH) team. We thank the research and clinical staff at the sites that participated in the Selenium and Vitamin E Prevention Trial (SELECT) study, without whom the trial would not have been successful. We are also grateful to the 35,533 dedicated men who participated in SELECT. The authors thank the Women’s Health Initiative (WHI) investigators and staff for their dedication, and the study participants for making the programme possible. A full listing of WHI investigators can be found at: http://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Short%20List.pdf
Data availability
Summary genetic association data for select cancer endpoints were obtained from the public domain: breast cancer (https://bcac.ccge.medschl.cam.ac.uk/bcacdata/); and overall prostate cancer (http://practical.icr.ac.uk/blog/). Summary genetic association data for colorectal cancer can be accessed by contacting GECCO (kafdem at fredhutch.org). Summary genetic association data on advanced prostate cancer can be accessed by contacting PRACTICAL (practical at icr.ac.uk). Summary genetic association data on type 2 diabetes from Vujkovic et al [34] (can be accessed through dbGAP under accession number phs001672.v3.p1 (pha004945.1 refers to the European-specific summary statistics). UK Biobank data can be accessed by registering with UK Biobank and completing the registration form in the Access Management System (AMS) (https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access).
Funding
JY is supported by a Cancer Research UK Population Research Postdoctoral Fellowship (C68933/A28534). JY, EEV, SJL, RMM and KKT are supported by Cancer Research UK (C18281/A29019) programme grant (the Integrative Cancer Epidemiology Programme) (https://www.cancerresearchuk.org/). AC acknowledges funding from a Medical Research Council PhD studentship (MR/N013794/1). EEV is supported by a Diabetes UK RD Lawrence Fellowship (17/0005587). EEV and CJB are supported by Diabetes UK (17/0005587) and the World Cancer Research Fund (WCRF UK), as part of the World Cancer Research Fund International grant programme (IIG_2019_2009). MV is supported by I01-BX003362 from VA Office of R&D. RMM is a National Institute for Health Research Senior Investigator (NIHR202411). RMM is also supported by the NIHR Bristol Biomedical Research Centre, which is funded by the NIHR (BRC-1215-20011) and is a partnership between University Hospitals Bristol and Weston NHS Foundation Trust and the University of Bristol. Department of Health and Social Care disclaimer: the views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. Disclaimer: where authors are identified as personnel of the International Agency for Research on Cancer/WHO, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer/WHO. This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration and was supported by award no. MVP000. This publication does not represent the views of the Department of Veterans Affairs, the US Food and Drug Administration, or the US government. This research was also supported by funding from the Department of Veterans Affairs awards I01- BX003362 (K-MC). The study sponsor/funder was not involved in the design of the study; the collection, analysis, and interpretation of data; writing the report; and did not impose any restrictions regarding the publication of the report. Funding specific to GECCO was from National Cancer Institute, NIH, US Department of Health and Human Services (U01 CA137088, R01 CA059045, U01 CA164930, R21 CA191312, R01201407). Genotyping/Sequencing services were provided by the Center for Inherited Disease Research (CIDR) contract number HHSN268201700006I and HHSN268201200008I . This research was funded in part through the NIH/National Cancer Institute (NCI) Cancer Center Support grant P30 CA015704. Scientific Computing Infrastructure at Fred Hutch was funded by ORIP grant S10OD028685. ASTERISK is a Hospital Clinical Research Program (PHRC-BRD09/C) from the University Hospital Center of Nantes (CHU de Nantes) and is supported by the Regional Council of Pays de la Loire, the Groupement des Entreprises Françaises dans la Lutte contre le Cancer (GEFLUC), the Association Anne de Bretagne Génétique and the Ligue Régionale Contre le Cancer (LRCC). The Alpha-Tocopherol, Beta-Carotene Cancer Prevention (ATBC) Study is supported by the Intramural Research Program of the US National Cancer Institute, NIH, Department of Health and Human Services. CLUE II funding was from the National Cancer Institute (U01 CA86308, Early Detection Research Network; P30 CA006973), National Institute on Aging (U01 AG18033) and the American Institute for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organisations imply endorsement by the US government. Cancer data from Maryland were provided by the Maryland Cancer Registry, Center for Cancer Prevention and Control, Maryland Department of Health, with funding from the State of Maryland and the Maryland Cigarette Restitution Fund. The collection and availability of cancer registry data are also supported by the Cooperative Agreement NU58DP006333, funded by the Centers for Disease Control and Prevention. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention or the Department of Health and Human Services. The work of ColoCare was supported by the NIH (grant numbers R01 [C.I. Li/C. M. Ulrich], U01 CA206110 [C. M. Ulrich/C. I. Li/E. M. Siegel/J. C. Figueiredo/G. A. Colditz], 2P30CA015704- 40 [G. Gilliland], R01 CA207371 [C. M. Ulrich/C. I. Li]), the Matthias Lackas-Foundation, the German Consortium for Translational Cancer Research, and the EU TRANSCAN initiative. The CCFR (www.coloncfr.org) is supported in part by funding from the National Cancer Institute (NCI), NIH (award U01 CA167551). Support for case ascertainment was provided in part from the Surveillance, Epidemiology, and End Results (SEER) Program and the following US state cancer registries: AZ, CO, MN, NC, NH; and by the Victoria Cancer Registry (Australia) and Ontario Cancer Registry (Canada). The CCFR Set-1 (Illumina 1M/1M-Duo) and Set-2 (Illumina Omni1-Quad) scans were supported by NIH awards U01 CA122839 and R01 CA143247 (to G. Casey. The CCFR Set-3 (Affymetrix Axiom CORECT Set array) was supported by NIH award U19 CA148107 and R01 CA81488 (to S. B. Gruber). The CCFR Set-4 (Illumina OncoArray 600K SNP array) was supported by NIH award U19 CA148107 (to S. B. Gruber) and by the Center for Inherited Disease Research (CIDR), which is funded by the NIH to the Johns Hopkins University, contract number HHSN268201200008I. Additional funding for the OFCCR/ARCTIC was through award GL201-043 from the Ontario Research Fund (to B. W. Zanke), award 112746 from the Canadian Institutes of Health Research (to T. J. Hudson), through a Cancer Risk Evaluation (CaRE) Program grant from the Canadian Cancer Society (to S. Gallinger), and through generous support from the Ontario Ministry of Research and Innovation. The SFCCR Illumina HumanCytoSNP array was supported in part through NCI/NIH awards U01/U24 CA074794 and R01 CA076366 (to P. A. Newcomb). The content of this manuscript does not necessarily reflect the views or policies of the NCI, NIH or any of the collaborating centres in the CCFR, nor does mention of trade names, commercial products or organisations imply endorsement by the US government, any cancer registry or the CCFR. The COlorectal cancer: Longitudinal, Observational study on Nutritional and lifestyle factors that may influence colorectal tumour recurrence, survival and quality of life (COLON) study is sponsored by Wereld Kanker Onderzoek Fonds, including funds from grant 2014/1179 as part of the World Cancer Research Fund International Regular Grant Programme, by Alpe d’Huzes and the Dutch Cancer Society (UM 2012–5653, UW 2013-5927, UW2015-7946), and by TRANSCAN (JTC2012-MetaboCCC, JTC2013-FOCUS). The NQplus study is sponsored as follows: by a ZonMW investment grant (98-10030); by the project PREVention of diabetes through lifestyle intervention and population studies in Europe and around the World (PREVIEW) project, which received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant no. 312057; by funds from TI Food and Nutrition (cardiovascular health theme), a public–private partnership on precompetitive research in food and nutrition; and by the Food Biomarker Alliance (FOODBALL), a project from JPI Healthy Diet for a Healthy Life. The CORECT Study was supported by the National Cancer Institute, NIH (NCI/NIH), US Department of Health and Human Services (grant numbers U19 CA148107, R01 CA081488, P30 CA014089, R01 CA197350; P01 CA196569; R01 CA201407; R01 CA242218), National Institutes of Environmental Health Sciences, NIH (grant number T32 ES013678) and a generous gift from Daniel and Maryann Fong. The Colorectal Cancer Study of Austria (CORSA) study was funded by Austrian Research Funding Agency (FFG) BRIDGE (grant 829675, to A. Gsur), the ‘Herzfelder’sche Familienstiftung’ (grant to A. Gsur) and was supported by COST Action BM1206. The American Cancer Society funds the creation, maintenance, and updating of the Cancer Prevention Study-II (CPS-II) cohort. This study was conducted with institutional review board approval. Colorectal Cancer Genetics & Genomics, Spanish study (CRCGEN) was supported by Instituto de Salud Carlos III, co-funded by FEDER funds –a way to build Europe– (grants PI14-613 and PI09-1286), Agency for Management of University and Research Grants (AGAUR) of the Catalan Government (grant 2017SGR723), Junta de Castilla y León (grant LE22A10-2), the Spanish Association Against Cancer (AECC) Scientific Foundation grant GCTRA18022MORE and the Consortium for Biomedical Research in Epidemiology and Public Health (CIBERESP), action Genrisk. Sample collection of this work was supported by the Xarxa de Bancs de Tumors de Catalunya sponsored by Pla Director d’Oncología de Catalunya (XBTC), Plataforma Biobancos PT13/0010/0013 and ICOBIOBANC, sponsored by the Catalan Institute of Oncology. We thank CERCA Programme, Generalitat de Catalunya for institutional support. The work of the Czech Republic CCS was supported by the Grant Agency of the Czech Republic (18-09709S, 20-03997S), by the Grant Agency of the Ministry of Health of the Czech Republic (grants AZV NV18/03/00199 and AZV NV19-09-00237) and by Charles University grants Unce/Med/006 and Progress Q28/LF1. The work of Darmkrebs: Chancen der Verhutung durch Screening (DACHS) was supported by the German Research Council (BR 1704/6-1, BR 1704/6-3, BR 1704/6-4, CH 117/1-1, HO 5117/2-1, HE 5998/2-1, KL 2354/3-1, RO 2270/8-1 and BR 1704/17-1), the Interdisciplinary Research Program of the National Center for Tumor Diseases (NCT), Germany, and the German Federal Ministry of Education and Research (01KH0404, 01ER0814, 01ER0815, 01ER1505A and 01ER1505B). The Diet, Activity, and Lifestyle Study (DALS) was supported by NIH (R01 CA48998 to M. L. Slattery). The work of the Early Detection Research Network (EDRN) is funded and supported by the NCI, EDRN Grant (U01 CA 84968-06). The coordination of EPIC is financially supported by the International Agency for Research on Cancer (IARC) and also by the Department of Epidemiology and Biostatistics, School of Public Health, Imperial College London, which has additional infrastructure support provided by the NIHR Imperial Biomedical Research Centre (BRC). The national cohorts are supported by the following bodies: Danish Cancer Society (Denmark); Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (Inserm) (France); German Cancer Aid, German Cancer Research Center (DKFZ), German Institute of Human Nutrition Potsdam-Rehbruecke (DIfE), Federal Ministry of Education and Research (BMBF) (Germany); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy, Compagnia di SanPaolo and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (the Netherlands); Health Research Fund (FIS) - Instituto de Salud Carlos III (ISCIII), Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, and the Catalan Institute of Oncology - ICO (Spain); Swedish Cancer Society, Swedish Research Council and County Councils of Skåne and Västerbotten (Sweden); and Cancer Research UK (14136 to EPIC-Norfolk; C8221/A29017 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk; MR/M012190/1 to EPIC-Oxford). (UK). The work of EPICOLON was supported by grants from Fondo de Investigación Sanitaria/FEDER (PI08/0024, PI08/1276, PS09/02368, P111/00219, PI11/00681, PI14/00173, PI14/00230, PI17/00509, 17/00878, PI20/00113, PI20/00226, Acción Transversal de Cáncer), Xunta de Galicia (PGIDIT07PXIB9101209PR), Ministerio de Economia y Competitividad (SAF07-64873, SAF 2010-19273, SAF2014-54453R), Fundación Científica de la Asociación Española contra el Cáncer (GCB13131592CAST), Beca Grupo de Trabajo ‘Oncología’ AEG (Asociación Española de Gastroenterología), Fundación Privada Olga Torres, FP7 CHIBCHA Consortium, Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR, Generalitat de Catalunya, 2014SGR135, 2014SGR255, 2017SGR21, 2017SGR653), Catalan Tumour Bank Network (Pla Director d’Oncologia, Generalitat de Catalunya), PERIS (SLT002/16/00398, Generalitat de Catalunya), CERCA Programme (Generalitat de Catalunya) and COST Action BM1206 and CA17118; CIBERehd is funded by the Instituto de Salud Carlos III.
The work of Epidemiological investigations of the chances of preventing, recognizing early and optimally treating chronic diseases in an elderly population (ESTHER)/VERDI was supported by grants from the Baden-Württemberg Ministry of Science, Research and Arts and the German Cancer Aid. In the Harvard cohorts, Health Professionals Follow-Up Study (HPFS) is supported by the NIH (P01 CA055075, UM1 CA167552, U01 CA167552, R01 CA137178, R01 CA151993, and R35 CA197735), Nurses’ Health Study (NHS) by the NIH (P01 CA087969, UM1 CA186107, R01 CA137178, R01 CA151993, and R35 CA197735), and Physicians’ Health Study (PHS) by the NIH (R01 CA042182). The Hawaii Adenoma Study is supported by NCI grant R01 CA72520. The Hwasun Cancer Epidemiology Study–Colon and Rectum Cancer (HCES-CRC) is supported by grants from Chonnam National University Hwasun Hospital, HCRI15011-1. In Kentucky, work was supported by a Clinical Investigator Award from Damon Runyon Cancer Research Foundation (CI-8) and grant NCI R01CA136726. The Leeds Colorectal Cancer Study (LCCS) was funded by the Food Standards Agency and Cancer Research UK Programme Award (C588/A19167). Melbourne Collaborative Cohort Study (MCCS) cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further supported by Australian NHMRC grants 509348, 209057, 251553 and 504711 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry (VCR) and the Australian Institute of Health and Welfare (AIHW), including the National Death Index and the Australian Cancer Database. Multiethnic Cohort (MEC) was supported by NIH grants (R37 CA54281, P01 CA033619 and R01 CA063464). The work of Molecular Epidemiology of Colorectal Cancer (MECC) was supported by the NIH, US Department of Health and Human Services (R01 CA081488, R01 CA197350, U19 CA148107, R01 CA242218) and a generous gift from Daniel and Maryann Fong. The work of Memorial Sloan Kettering Cancer Center (MSKCC) at Sloan Kettering in New York was supported by the Robert and Kate Niehaus Center for Inherited Cancer Genomics and the Romeo Milio Foundation. Moffitt was supported by funding from the NIH (grant numbers R01 CA189184, P30 CA076292), Florida Department of Health Bankhead-Coley Grant 09BN-13, and the University of South Florida Oehler Foundation. Moffitt contributions were supported in part by the Total Cancer Care Initiative, Collaborative Data Services Core and Tissue Core at the H. Lee Moffitt Cancer Center & Research Institute, a National Cancer Institute-designated Comprehensive Cancer Center (grant number P30 CA076292). We acknowledge funding support for NCCCS I & II from the NIH, R01 CA66635 and P30 DK034987. This work of Newfoundland Colorectal Cancer Registry (NFCCR) was supported by an Interdisciplinary Health Research Team award from the Canadian Institutes of Health Research (CRT 43821), the NIH, US Department of Health and Human Services (U01 CA74783), and National Cancer Institute of Canada grants (18223 and 18226). The authors wish to acknowledge the contribution of A. Belisle and the genotyping team of the McGill University and Génome Québec Innovation Centre, Montréal, Canada, for genotyping the Sequenom panel in the NFCCR samples. Funding was provided to M. O. Woods by the Canadian Cancer Society Research Institute. The Northern Swedish Health and Disease Study (NSHDS) research was supported by Biobank Sweden through funding from the Swedish Research Council (VR 2017-00650, VR 2017-01737), the Swedish Cancer Society (CAN 2017/581), Region Västerbotten (VLL-841671, VLL-833291), Knut and Alice Wallenberg Foundation (VLL-765961), and the Lion’s Cancer Research Foundation (several grants) and Insamlingsstiftelsen, both at Umeå University. Ohio State University Medical Center (OSUMC) Ohio Colorectal Cancer Prevention Initiative (OCCPI) funding was provided by Pelotonia and HNPCC funding was provided by the NCI (CA16058 and CA67941). The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial and the Intramural Research Program of the Division of Cancer Epidemiology and Genetics were supported by contracts from the Division of Cancer Prevention, National Cancer Institute, NIH, DHHS. Funding was provided by NIH, Genes, Environment and Health Initiative (GEI) Z01 CP 010200, NIH U01 HG004446 and NIH GEI U01 HG 004438. For SEARCH, the University of Cambridge has received salary support in respect of PDDP from the NHS in the East of England through the Clinical Academic Reserve. Cancer Research UK (C490/A16561) and the UK National Institute for Health Research Biomedical Research Centres at the University of Cambridge also provided support. SELECT research reported in this publication was supported in part by the National Cancer Institute of the NIH under Award Numbers U10 CA37429 (C. D. Blanke) and UM1 CA182883 (C. M. Tangen/I. M. Thompson). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The work of Screening Markers For Colorectal Disease Study and Colonoscopy and Health Study (SMS-REACH) was supported by the National Cancer Institute (grant P01 CA074184 [to J. D. Potter and P. A. Newcomb], grants R01 CA097325, R03 CA153323 and K05 CA152715 [to PAN]) and the National Center for Advancing Translational Sciences at the NIH (grant KL2 TR000421 [to ANB-H]). The Swedish Low-risk Colorectal Cancer Study was supported by grants from the Swedish Research Council (K2015-55X-22674-01-4, K2008-55X-20157-03-3, K2006-72X-20157-01-2) and the Stockholm County Council (ALF project). The Swedish Mammography Cohort and Cohort of Swedish Menwas supported by the Swedish Research Council /Infrastructure grant, the Swedish Cancer Foundation, and the Karolinska Institute´s Distinguished Professor Award to A. Wolk. UK Biobank research has been conducted using the UK Biobank Resource under Application Number 8614. VITamin D and OmegA-3 TriaL (VITAL) was supported by NIH (K05 CA154337). The Women’s Health Initiative (WHI) program is funded by the National Heart, Lung and Blood Institute, NIH, US Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C and HHSN271201100004C.
Authors’ relationships and activities
DG is employed part-time by Novo Nordisk for work unrelated to that presented here. All other authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
JY, EB, DG and KKT contributed to study conception and design, acquisition of data, analysis and interpretation of data, drafting the article and revising it critically for important intellectual content, and gave final approval of the version to be published. AC contributed to analysis of data, drafting the article and revising it critically for important intellectual content, and gave final approval of the version to be published. MV contributed to acquisition of data, drafting the article and revising it critically for important intellectual content, and gave final approval of the version to be published. CJB, EEV, KB, RMM, OD, SJL, VM, KC, BFV, PST, MJG, JH, AJP, PDPP, RES, SG, MAJ and RKP contributed to study design, drafting the article and revising it critically for important intellectual content, and gave final approval of the version to be published. JY is the guarantor of this work.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Dipender Gill and Kostas K. Tsilidis are joint senior authors and contributed equally to this work.
Membership of the PRACTICAL consortium is provided in the electronic supplementary material (ESM). Further information on the PRACTICAL consortium can be found at http://practical.icr.ac.uk/.
Supplementary Information
Below is the link to the electronic peer-reviewed but unedited supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yarmolinsky, J., Bouras, E., Constantinescu, A. et al. Genetically proxied glucose-lowering drug target perturbation and risk of cancer: a Mendelian randomisation analysis. Diabetologia 66, 1481–1500 (2023). https://doi.org/10.1007/s00125-023-05925-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-023-05925-4